

Q&As
Q&As
Möchten Sie die jeweilige Frage und Antwort in Ihrer Sprache suchen? Ändern Sie die Sprache in der Auswahlliste oben.
They will be available on our website in PACT or via the assessment of regulatory needs list.
You can see from the ECHA PACT website or via the assessment of regulatory needs list if there is a group assessment report for your substance. If yes, you can download ECHA’s report on the assessment of regulatory needs, which includes a list of substances within this group. Note that the assessment is done within a short time period; it is iterative and for most of the groups an updated report will be published for instance after the generation of additional hazard information, where needed. Note also that this is preparatory work for the existing formal regulatory processes; any assessment in one of the formal processes will be communicated on ECHA’s website as usual in accordance with the respective procedures.
No, ECHA will not publish a list of the groups as such and their member substances. ECHA will publish the assessment of regulatory needs done on each group and ECHA’s reports include the list of the substances covered in each group. They will be available on our website in PACT or via the Assessment of regulatory needs list.
For its assessments of regulatory needs, ECHA forms and selects groups according to several criteria, including among others the known or suspected hazards of some group members, or the position of the substances in the chemical universe [see also Q&A 1869]. Normally, it is not foreseen that ECHA forms and selects groups on request; in case of concrete suggestions/ questions, however, please contact ECHA.
In most cases, compliance check is proposed as the first action and when that has started for a substance you find further information on the status under Dossier Evaluation on ECHA’s website. In case a compliance check has been opened for your registration, ECHA also informs you via REACH-IT.
PACT will also show when any of the other REACH/CLP processes has started, and also sometimes when an authority has submitted an intention (e.g. for harmonised classification).
Registrants can find ECHA’s group assessment reports on the ECHA PACT website or via the assessment of regulatory needs list. This report gives an overview of the assessment for the group, including if there is (no) action planned for the moment for any substance within the group. When ECHA updates the assessment, a new report will be published.
The initial grouping is done primarily using IT-based algorithms and following two broad, complementary methods:
- structural similarity, which uses the substance identity information in registration dossiers and C&L notifications; and
- associations made by the registrants between substances through read-across and category approaches as well as category associations from external sources (e.g. OECD categories).
The algorithmically generated groups are verified by chemists and group membership is reconsidered through the group assessment.
Structurally similar substances are identified within the universe of registered substances around pre-selected substances known as ‘seeds’. Examples of seeds are substances in Annex VI to the CLP Regulation, in the Candidate List or listed in the CoRAP, for which there is already an identified or potential hazard. Another starting point for grouping could be a substance that has a certain type of use or function with a potential for exposure.
Note that these methods are different from grouping as defined in Section 1.5 of Annex XI to REACH and therefore do not constitute validated read-across and category information according to that requirement.
ECHA utilises all available information for grouping substances. The algorithms use all available identifiers in all registrations, compositions and reference substances in IUCLID dossiers. Hence, it is quite likely that all structurally related substances are identified, to the extent that the submitted information allows to do so. However, for certain substances, such as inorganic UVCBs, substance grouping may be less accurate and may require additional manual effort for fine tuning the approach and manually verifying the accuracy of the results.
Substance grouping by ECHA is designed to be inclusive and is robust to the extent that is possible. E.g. small differences in molecular structure that are considered insignificant for grouping can be toxicologically relevant. The assessment of the regulatory needs of the group may include such considerations, in which case it is possible that the group will be split in smaller subgroups, or the substances will be assessed individually. Hence, the robustness of the group depends on the process/stage we are at and at the level of certainty this stage requires. Therefore, group membership is reconsidered throughout the iterative group assessments.
Groups are formed based on similarity of chemical structures and cover both substances with known/ likely hazards and substances not currently known or suspected to be hazardous. ECHA may address (potentially) hazardous substance earlier than substances with unlikely hazards, but the systematic grouping of the chemical universe will address all REACH registered substances eventually.
Registrants cannot influence ECHA’s initial grouping based on structural similarity. The purpose of ECHA’s grouping based on structural similarity is mainly to facilitate and speed up the assessment of substances. However, up-to-date information in registration dossiers will be taken into account during any potential follow-up regulatory action as well as at later iterations of the assessment of regulatory needs of the group. Also any changes in read-across or category adaptations by the registrants will be taken into account by ECHA in later iterations of the assessment of regulatory needs.
See also Q&A 1878 on the benefits of updating registration dossiers.
ECHA’s assessment of regulatory needs is mainly based on the information provided in the REACH registration dossiers. A (foreseen) need for regulatory risk management under REACH, CLP and other EU legislations depends on the potential hazards identified, the uses and potential for exposure or releases identified. The assessment of regulatory needs is an iterative process that can start from a low level of information and high uncertainty on the best way forward, moving to data generation and to proposing more definitive regulatory management options for the (groups of) substance(s). Note that the assessment of regulatory needs is iterative: the foreseen regulatory actions may therefore change based on new information, further assessment or change in policy.
EU regulatory risk management actions are expected for many assessed substances. However, most of these substances may require further data generation and confirmation of their hazards before the need for planned actions can be confirmed or actions can be initiated.
Regulatory risk management actions can be initiated either by Member States and/or ECHA (upon request by the Commission) through the formal processes.
ECHA is mainly using the information on uses (e.g. life cycle stages, type of products, use description) reported in the REACH registration dossiers (IUCLID) as a proxy for assessing the potential for exposure to humans and releases to the environment. The potential for release/exposure is generally considered high for “widespread” uses, i.e. professional and consumer uses and uses in articles. For these uses, normally happening at many places, the expected level of control is, in theory, considered limited. No quantitative exposure assessment is performed at this stage and so the chemical safety reports are normally not consulted. In some cases, public information can also be used.
The potential for exposure is then considered together with the (potential) hazard to assess the regulatory needs for the (group of) substance(s).
The initial grouping by ECHA provides a useful starting point for assessing substances. However, it does not necessarily mean that the same regulatory needs will be identified for all substances within a group or that the full group will be progressed towards the same regulatory action by authorities. Actions proposed as outcome of our assessments can be on the entire group, subgroup(s), or individual substance(s). For instance, ECHA may propose restriction or CLH for one substance, a sub-set of substances in the group or for the entire group.
We take ongoing actions into account in the assessment of regulatory needs of the group however the assessment will not impact the ongoing regulatory actions. We check whether there are any planned or ongoing activities with the substances in the group before the assessment of regulatory needs is done on the group.
A formal assessment of registrant’s read-across /category approach is only done in the context of the relevant formal processes (e.g. compliance check); ECHA does not formally accept or confirm the registrant’s read-across in its assessments of regulatory needs. See also Q&A 1868 on how the grouping is done by ECHA.
When "currently no need for further EU RRM action" is proposed based on available information, this may be because of several considerations e.g. on hazard, registration status, existing legislations, uses, potential for exposure or combinations of those. Therefore, if new information becomes available (e.g. on hazard, uses) this conclusion will need to be revisited.
A conclusion on “Currently no need for further EU RRM action” based on the current registered uses and hazard information doesn’t necessarily mean the substance is safe for all future new uses. In addition, in some specific situations where “Currently no need for further EU RRM action” is foreseen, reference is made to company level risk management and the responsibility of registrants and downstream users to ensure safe use of the substances in any case. For instance, registrants and downstream users are expected to adequately (self) classify their substances and implement the necessary risk management measures and operational conditions to ensure the safe use of the substances.
One of the aims of the publication of the ECHA group assessment reports is to increase transparency of authorities' work in the early stages of the assessment and to give registrants the possibility to clarify the potential hazard and use profile of their substances. The improved quality of the registration dossiers will in turn provide authorities a more solid basis for deciding on the need for further actions. Therefore, if we have in registration dossiers accurate information on hazards and uses this will impact the way substances are proceeding to further processes.
It is in the interest of both authorities and industry that we focus on hazardous substances for which uses are of relevance from a regulatory risk management perspective.
If you consider that the information in your registration is not up-to-date, or that you have additional data that may assist with the assessment of your substance, ECHA recommends updating your dossier as soon as feasible for you. The updated information will be taken into account during the ongoing or future group assessment or during any potential follow-up regulatory action. In any case, you as a registrant have the obligation to keep your dossiers up to date with the latest available information.
In the context of the assessments of regulatory needs of groups of substances, ECHA does not foresee direct interaction with the registrants. We highly recommend that registrants update their registrations proactively on hazards, uses and volumes information as soon as possible and preferably before the planned regulatory actions would start. The information provided in the update will be taken into account by ECHA in the following formal processes and when updating the assessment of regulatory needs. In case of additional questions we would recommend to contact ECHA.
Once an assessment of regulatory needs for a group of substances is published, we encourage registrants to consult them and to consider the concern identified (if any) and the proposed next steps.
Whenever a formal process is initiated on a substance, the information is available on our website, along with the contact details of the authority initiating the action. We also encourage industry to contribute to the consultations foreseen in the different regulatory processes.
Yes. The List of Substances Subject to Authorisation (Annex XIV of the REACH Regulation) includes several substances.
Substances are regularly added to Annex XIV by the European Commission, on the basis of recommendations issued by ECHA. The link to the updated Annex XIV can be found on ECHA's website at: https://echa.europa.eu/authorisation-list.
Further details on the procedure for the inclusion of substances to Annex XIV of the REACH Regulation are available in FAQ ID=127, as well as on ECHA's website at: http://echa.europa.eu/regulations/reach/authorisation.
The Candidate List of Substances of Very High Concern (SVHC) for authorisation (Candidate List) is available on ECHA's website at: http://echa.europa.eu/candidate-list-table.
Additional substances are regularly included in the Candidate List, once these have been identified as SVHC.
Substances included in the Candidate List may be prioritised for inclusion in Annex XIV of the REACH Regulation (the so called "Authorisation List"). The Authorisation List contains all substances which, after a certain deadline, may only be used and/or placed on the market after a specific authorisation has been granted.
ECHA has to make at least every second year a recommendation of priority substances for inclusion in Annex XIV to the European Commission. Interested parties are invited to submit comments during this process. In addition, the Member State Committee issues an opinion on the recommendation before it is submitted to the European Commission. The European Commission then decides using the comitology procedure which of the recommended substances are to be included in Annex XIV and specifies, based on ECHA's recommendation, the transitional arrangements and, where relevant, exemptions and review periods. Further details on the procedure for inclusion of substances in Annex XIV of the REACH Regulation are available on ECHA's website at http://echa.europa.eu/addressing-chemicals-of-concern/authorisation/recommendation-for-inclusion-in-the-authorisation-list .
Applications for authorisation need to be made within the deadline (the so called "latest application date") that is specified in the "Authorisation List" for the corresponding substance if the applicant wishes to use the substance without interruption after the sunset date. Authorisation applications need to be submitted to ECHA. Third parties can provide information on alternative substances and technologies during public consultations on the uses that authorisation has been applied for. These are made available on ECHA's web-site. The ECHA Committees for Risk Assessment (RAC) and Socioeconomic Analysis (SEAC) give draft opinions on the application. Applicants will have the opportunity to comment on these draft opinions. RAC and SEAC will adopt final opinions and ECHA sends them to the European Commission. The European Commission decides, using the comitology procedure, whether an authorisation is granted or refused. ECHA will establish a publicly available database that will contain summaries of the Commission decisions. Further details on the application for authorisation procedure are available on ECHA's website under the following links:http://echa.europa.eu/regulations/reach/authorisation http://echa.europa.eu/addressing-chemicals-of-concern/authorisation/applications-for-authorisation
Applications for authorisation may be submitted in any one of the official EU languages selected by the applicant. This means that the whole application, including the attachments and the Chemical Safety Report, must be submitted in the same language. This is a legal requirement based on Article 104(1) of REACH and Article 2 of Regulation No 1 of 15 April 1958.
Only manufacturers, importers or downstream users of an Annex XIV substance as well as duly mandated Only Representatives can apply for an authorisation and be holders of a granted authorisation.
Yes. A duly mandated Only Representative ("OR") of a non-EU manufacturer can apply for an authorisation regardless of whether the OR assisted the non-EU entity with the registration of the Annex XIV substance to date. Once the OR has been appointed, it will have to comply with all other applicable obligations under REACH on behalf of the non-EU manufacturer who has appointed them. In cases where a non-EU manufacturer has not yet been appointed an OR, for example due to no obligation to register a substance, an OR may nevertheless be appointed. The OR will then represent the non-EU manufacturer with regard to all applicable REACH obligations.
The holder of an authorisation is the person who submitted the authorisation application to ECHA. However, a downstream user may continue his use of an Annex XIV substance provided that this use is in accordance with the conditions of an authorisation granted to an actor up his supply chain for that use. Moreover, a manufacturer, importer or downstream user can continue placing an Annex XIV substance on the market for a use for which his immediate downstream user was granted an authorisation.
The European Commission is responsible for taking decisions on Applications for authorisations. ECHA's Committees for Risk Assessment (RAC) and Socio-Economic Analysis (SEAC) will adopt opinions on the application for authorisation which will be taken into account by the Commission in its final decision.
Yes. Be aware, however, that ECHA recommends applicants to submit their applications within submission windows that are published on its website. This concept ensures a batch wise process and a good synchronisation with the operational schedule of RAC and SEAC committees.
You should apply before the Latest Application Date to be sure that you will be able to continue to use the substance while your application is being treated by ECHA and the Commission. In other words, you should apply early to take advantage of the transitional arrangements and continue your use after the sunset date even if no decision has been taken by the Commission.
The date of submission of the application will be considered as the date on which your application has been received for the purpose of benefitting from the transitional arrangements described in Article 58(1)(c)(ii), provided that you pass the business rules checks. This date is relevant if you intend to take advantage of these transitional arrangements and continue your use after the sunset date if no decision has been taken by the Commission. The following two situations may occur:
1. You have submitted your application early enough and it passes business rules checks before the latest application date: in this case you will benefit from the transitional arrangements described in Article 58(1)(c)(ii) provided that you pay your invoice in due time.
2. You have submitted your application just before the latest application date but it passes the business rules after the latest application date: in this case you can benefit from the transitional arrangements described in Article 58(1)(c)(ii) provided that you pay your invoice in due time. If your application does not pass the business rules checks you will have to resubmit your application. If you resubmit your application after the latest application date you will not be able to benefit from the transitional arrangements.
Therefore ECHA recommends that you submit your application during the submission window three months earlier than the latest application date. If you choose to submit in the latest submission window, ECHA recommends that you do so at the very beginning of the window.
The date from which the 10 months' time limit for the Committees to prepare their draft opinions starts is the date on which ECHA has received the full application fee.
Yes, the application will still be accepted for processing and evaluated by RAC and SEAC. However, the transitional arrangements under Article 58 (1)(c)(ii) of the REACH Regulation will not apply. These transitional arrangements would allow the applicant to use the substance even after the Sunset Date if no decision has been taken by the Commission.
If the substance has a threshold you need to demonstrate that the risks associated for the hazard endpoint mentioned in Annex XIV are adequately controlled. If the substance does not have a threshold, you need to demonstrate minimisation of emissions and exposure as far as possible. The Chemical Safety Report (CSR) should therefore focus on the Annex XIV endpoint but information on other endpoints might be necessary for comparing the risks with the alternatives. The alternatives should result in reduced overall risks to human health and the environment. Therefore, it is important not only to consider the risks arising from the Annex XIV endpoint but also on all other possible risks from the Annex XIV substance and the alternatives.
A decision on authorising a use can be different from one use to another. Thus, if certain uses are authorised and others are not, then the non-authorised uses are no longer permitted.
The primary objective of RAC and SEAC is to provide consistent opinions of high scientific quality to support the desicion making of the European Commission. The remits of both Committees are clear and cooperation between them is ensured by agreed procedures.
See also: http://echa.europa.eu/documents/10162/13555/common_approach_rac_seac_en.pdf
No. A distributor who only stores the Annex XIV substance before placing it on the market cannot be considered as Downstream Users in the context of Article 56(1)(e) (see Article 3(14) of the REACH Regulation). However, actors can no longer be considered as distributors if they use the substances themselves (e.g. repackaging). Then they are considered as downstream users and, as such, shall apply for authorisation (and may cover uses of their downstream users), unless already covered by an authorisation.
Distributors (i.e. entities who only store the substance) are "transparent" in the supply chain if they do not "use" the substance. Therefore an application granted to a downstream user should be understood as covering the manufacturer/importer of that substance and all the distributors in between. However, this only applies in cases where there are no actors using the substance (e.g. formulators, repackaging companies, etc.) between the downstream user applicant and the manufacturer/importer. Also, distributors have to communicate the relevant information (e.g. safety data sheets, authorisation numbers) to their downstream users.
No. A downstream user (Company B) holding an authorisation for his use(s) (for instance, an end-use) can be supplied by an actor (Company A) up his supply chain (for instance, a formulator) in accordance with Article 56(1)(e). However, the use of Company A (that is the formulation of the Annex XIV substance) cannot be covered by an authorisation granted down the supply chain to Company B. Company A needs to have his use (the formulation) covered by a separate authorisation granted directly to him or to an actor up his supply chain (for instance the manufacturer of the Annex XIV substance). Note that distributors who only store the Annex XIV substance before placing it on the market cannot be considered as Downstream Users in the context of Article 56(1)(e), see Q&A 577 for more details.
Information presented should be for the substance as the authorisation is (or is not) granted for a given substance.
See also FAQ ID=130 in Frequently Asked Questions about REACH.
Yes. An authorisation can be reviewed before the expiry of the review period. REACH Art 61(2) specifies that: "Authorisations may be reviewed at any time if: (a) the circumstances of the original authorisation have changed so as to affect the risk to human health or the environment, or the socio- economic impact; or (b) new information on possible substitutes becomes available"". In this case, the Commission shall set a reasonable deadline by which the holder(s) of the authorisation may submit further information necessary for the review and indicate by when it (i.e. the Commission) will take a final decision.
An applicant has the right to contest the decision of the Commission before the General Court.
The enforcement of REACH is a responsibility of each EU Member State, as well as the members of the EEA (Norway, Iceland and Liechtenstein). They must ensure that there is an official system of controls and lay down legislation specifying penalties for non-compliance with the provisions of REACH. See also FAQ ID=3 in Frequently Asked Questions about REACH.
All information available to ECHA could be used; for instance, CLP notifications, dossier evaluation results or any relevant information arising from the public consultation on alternatives.
It will take about 2 years. Article 64 of the REACH Regulation gives many details. Below we give an overall description of the timelines. Once you have submitted the application it takes about 2-3 months for its processing and for the application fee to be received by ECHA. The Committees will prepare their draft opinions within 10 months from that date of receipt of the fee. The applicant can comment on the draft opinions within 2 months before the Committees adopt their final opinions. This will take 2 months. Some weeks are also reserved for sending and receiving the draft opinion. Thus, it takes about 17-18 months for the applicant to receive the final opinions from the date it had submitted the application. ECHA will publish the opinions on its website and send them to the Commission, the Member States and the applicant. A final decision granting or refusing the authorisation shall be taken via a 'comitology' procedure (see the "examination procedure" referred to in Article 5 of Regulation (EU) No 182/2011). All in all, it would normally take about 6 months to have the final decision from the publication of the opinions of the Scientific Committees on ECHA's website.
The criteria for granting an authorisation are clearly defined in Art. 60 of REACH: under the "adequate control route" (Art. 60(2)) an application shall be granted if the risk to human health and the environment from the use of the substance arising from the intrinsic properties specified in Annex XIV is adequately controlled.
Under the "socio-economic route" (Art. 60(4)), an authorisation may be granted if it is shown that (i) the socio-economic benefits outweigh the risk to human health and the environment from the use of the substance and (ii) there is no suitable alternative substances or technologies.
The factors to be taken into account for assessing the availability of suitable alternatives are described in Art. 60(5) and in the Guidance on Applications for Authorisation.
Also, two important documents describe how RAC and SEAC intend to evaluate the applications:
No. An authorisation number is unique to each combination of [applicant-substance-use applied for]. If the downstream user is himself an applicant (in either single or joint application), he will get its own authorisation number(s) related to the authorised use(s). If the downstream user is not an applicant but that he relies on an authorisation granted to a manufacturer/importer up his supply chain for his uses, the downstream user will not receive his own authorisation number(s) but he will be informed by his supplier about the authorisation number (which should be mentioned at least on the label of the product).
Yes. After the sunset date, according to Article 69(2), ECHA has the obligation to consider the risks related to the corresponding Annex XIV substance in articles and possibly apply the restriction procedure if the risk to human health or the environment is not adequately controlled. In this case, the Agency shall prepare a dossier which conforms to the requirements of Annex XV. This assessment will have to be done for each Annex XIV substance after its corresponding sunset date.
In addition, Member States or ECHA (on request from the Commission) may propose a restriction at any time on the use of any substance (including SVHCs) in articles if the risks arising from the use of these articles are not properly controlled.
Yes. The non-EU companies can also be located in different jurisdictions. The application fee will be assessed for each non-EU legal entity the Only Representative represents in the application.
Only Representatives have to sign-up in REACH-IT for each non-EU company they represent and submit the relevant IUCLID dossier using the appropriate accounts. It is not possible to use the same Legal Entity Object (having the same company UUID) for multiple accounts, but it is possible to use the same company identification information (name, VAT, etc.). Similarly as for Registration, Only Representatives must indicate in the "company size" field of REACH-IT the size of the non-EU company they are representing. In the determination of their size, linked and partner enterprises to the company the Only Representative represents should also be taken into account.
In addition, Only Representatives are advised to attach clear documentation of their appointment in their application (for instance, a copy of the appointment) in the field "Assignment from non EU manufacturer" of the IUCLID file (IUCLID section 1.7). Only Representatives are also advised to indicate the list of the importers' names covered by the application in the field "Other importers".
For more details:
No. ECHA will not inform the applicant of its decision on all the confidentiality claims in the application for authorisation.
On the other hand, there may be situations where ECHA finds it necessary to make publicly available information that the applicant claimed confidential. In such cases ECHA will inform the applicant of this decision and allow him to respond appropriately.
An example of such a situation is if insufficient information falling under the broad information of uses is given in the public parts of the application for authorisation ("public versions" of the assessment reports, i.e. the Chemical Safety Report, Analysis of alternatives, Socio-economic analysis, Substitution plan or their annexes). In such a case ECHA has reserved the right under Article 64(2) of REACH to supplement the broad information package for the public consultation on alternatives with the necessary information from the "complete versions" of these assessment reports with the aim to make the public consultation meaningful. ECHA gives the applicant the opportunity to comment its proposal for broad information of uses before the final version is issued and published.
ECHA has consulted the services of the European Commission on this. As a general principle, if you apply for authorisation before the latest application date, the review period would be counted from the sunset date. In practice, this means that applying early would not shorten your time for placing on the market and/or using the substance after the sunset date as compared to applying closer to the latest application date.
If you apply for authorisation after the latest application date, the placing on the market and use of the substance will no longer be allowed as from the sunset date, unless a decision granting an authorisation has been adopted by then. If the decision is adopted before the sunset date, the review period would in principle be counted from the sunset date. If a decision is adopted after the sunset date, the review period would be counted from the date of entry into force of the decision
a) Do I have to stop using the substance?
b) Do I have to notify my use to ECHA?
a) It depends.
You do not have to stop if the application made up your supply chain has been submitted before the substance’s latest application date. In this case, you can continue using the substance after its sunset date, awaiting for the European Commission’s decision.
You have to stop if the authorisation application has been submitted after the substance’s latest application date. You can restart using the substance only if the European Commission decides to grant an authorisation for your use.
b) Not yet. As long as the European Commission’s decision on the authorisation application covering your use is pending, you cannot notify your use to ECHA. The notification of authorised uses is possible only after an authorisation has been granted. Once the decision to grant an authorisation has been adopted, you then need to submit a downstream user notification of your authorised use (Article 66 of REACH) to ECHA within three months from the first delivery of the substance (see also Q&A 1441).
Yes. If you rely on an authorisation granted to an actor up your supply chain, you must notify your authorised use to ECHA. You must submit your notification within three months from the first delivery of the substance following issuing of the authorisation decision. Your notification needs to refer to the relevant authorisation number(s). Authorisation numbers are indicated on the label of the product and the safety data sheet you receive from your supplier. As they are use-specific, you need to select the specific authorisation number(s) which correspond to your use. Authorisation numbers have the format 'REACH/x/x/x' (see also Q&A 750).
Naturally, you need to comply with the conditions of the authorisation, which should be communicated to you by your supplier in the safety data sheet.
For further information on the downstream user notification for authorised uses (Article 66 of REACH), see:
https://echa.europa.eu/support/dossier-submission-tools/reach-it/downstream-user-authorised-use
ECHA will publish the emissions of non-threshold substances such as environmental endocrine disruptors, PBTs, and vPvBs. It will also publish the release factors related to these emissions to the environment in cases where the applicant has not claimed the volumes used to be confidential. In such cases, ECHA will disclose the emissions as such and redacted information on the release factor (see example).
ECHA encourages applicants to be transparent about the release factors. Indeed, in two thirds of applications received to date (March 2020), applicants have provided this information without claiming the information on the release factor and thus the volume used confidential.
Note that the actual release factors and volumes used are always made available to ECHA’s Committees for Risk Assessment and Socio-economic Analysis when they evaluate the applications and give opinions to the European Commission.
Justification:
Article 64(6) of the REACH Regulation says that the Agency shall determine in accordance with Article 118 and 119 which parts of its opinions should be made publicly available on its website. Article 118(2)(c) provides that disclosure of precise tonnage is normally deemed to undermine the protection of commercial interests of the concerned party.
Under the Aarhus Regulation (Article 6(1)), an overriding public interest in disclosure is deemed to exist where a request is made for information relating to emissions to the environment. ECHA thus considers that emission values are non-confidential from a viewpoint of public interest and right to know.
This policy is consistent with the ECHA’s core values of transparency and efficiency, recognising the overriding public interest in the disclosure of emissions set out in the Aarhus Regulation.
Example:
The volume used of substance is 2 000 kg per year, the release factor is 0.2% and thus the emissions to the environment are 4 kg per year. Unless the applicant has claimed the volume confidential, ECHA will publish this information as part of its opinion.
If the applicant claimed that the volume used should not be disclosed to the public, ECHA would instead publish the volume within a range (e.g. 1 000-5 000 kg), the corresponding release factor also within a range (e.g. 0.1%-0.4 %) and the emission to the environment (4 kg).
No. The REACH Regulation does not provide for the possibility to grant a ‘grace period’ or transitional arrangements. This means that as of the date of the notification of the Commission decision to the authorisation applicant(s) the non-authorised use of the substance will need to be stopped immediately by both the applicant(s) and any downstream users.
Authorisation is required for the use of a substance included in Annex XIV of REACH, either on its own or in a mixture. If a substance is used as such, this is a use of the substance on its own, and the exemptions in Article 56(6) (a) and (b) of REACH cannot be applied. If the substance is used in a mixture, then the aforementioned exemptions may be applied. These exemptions apply to SVHCs meeting the criteria of points (d), (e) and (f) of Article 57 of REACH below a concentration limit of 0.1 % w/w, and for all other substances below the values specified in Article 11(3) of Regulation (EC) No 1272/2008 which result in the classification of the mixture as hazardous.
The authorisation requirement does not apply to the use of CMR substances when they are present in mixtures below the concentration limits specified in Article 11(3) of the CLP Regulation, which result in the classification of the mixture as hazardous (Article 56(6)b of REACH).
Only the hazard class (or hazard classes) which specifically led to inclusion of the substance in the authorisation list should be taken into account.
The basis for this is to target the intrinsic properties of highest concern, which is the objective of the authorisation process.
This is reflected in the legal provisions of REACH:
- Article 62(4), which requires an application for authorisation only for the hazard class given in the authorisation list, and
- Article 60(2), which limits the assessment of a request for authorisation to the hazard class given in the authorisation list under the adequate control route.
No. There is no tonnage threshold below which (the placing on the market for) a use of an Annex XIV substance is exempted from the authorisation requirement.
The authorisation requirement applies to the placing on the market and use of a substance on its own as listed in Annex XIV. Therefore, it usually does not apply if the Annex XIV substance is only an impurity or additive or constituent of another substance, unless this is specified in the Annex XIV entry (e.g. substance W and substances X, Y and Z containing substance W in a concentration ≥ x %) or the other substance is also listed in Annex XIV.If a substance listed in Annex XIV is included as a component in a mixture, the authorisation requirement applies for this use (i.e. the formulation of the mixture). Further, the placing on the market and use of such mixtures require authorisation, unless the Annex XIV substance is present in the mixture below the concentration limits set out in REACH Article 56(6).
See also: Guidance for identification and naming of substances under REACH (link to http://echa.europa.eu/documents/10162/13643/substance_id_en.pdf)
No. Uses of recovered substances are not exempted from the authorisation requirement. As in case of use(s) of any other Annex XIV substance, use(s) of a recovered substance whose identifiers correspond to those of an entry in the Annex XIV of REACH is subject to the authorisation requirement, unless the use of the substance is specifically exempted otherwise.
See also: Guidance on waste and recovered substances http://echa.europa.eu/documents/10162/13632/waste_recovered_en.pdf
Yes. Under Article 3(23) of REACH, scientific research and development means any scientific experimentation, analysis or chemical research carried out under controlled conditions in a volume less than one tonne per year. Thus, the use of an Annex XIV substance in analysis is exempted from authorisation under Article 56(3) if the substance is used, on its own or in a mixture, in analytical activities such as monitoring and quality control where these activities are carried out under controlled conditions and in a volume not exceeding one tonne per year and per legal entity.
The exemption applies to the use of an Annex XIV substance when it is required as part of an analytical method for the measurement of another substance or property (e.g. used as an extraction solvent or reagent, or to validate the technical specification or performance of a product) and the analysis of the Annex XIV substance itself (e.g. for quality or process control). Where these conditions are met, there is no need to apply for an authorisation for this use or to include this task as a working contributing scenario (i.e. PROC 15) in an application for authorisation.
This exemption applies irrespective of where the analysis is performed i.e. on-site or off-site facilities. However, this exemption does not cover sampling activities (see Q&A 1153).
- Yes, these exemptions cover the incorporation of a substance into the product during the manufacturing process.
- Yes, the uses of a substance upstream preceding an exempted end-use are also exempted but only in the volumes ending up in the exempted end-use. It should be noted that, with regard to uses in cosmetic products and in food contact materials, the exemption only applies when the intrinsic properties specified in Annex XIV for the substance in question concern hazards to human health.
Yes, the uses of a substance upstream, preceding "use as fuels in closed systems", are also exempted under the condition that the control of the risks – i.e., use in closed systems – is also pursued in the upstream life-cycle steps preceding the end-use as a fuel.
Pursuant to Articles 60(2) and 62(6) of REACH, an application for authorisation is not required for a substance used in a medical device regulated under Directives 90/385/EEC, 93/42/EEC or 98/79/EC if that substance has been identified in Annex XIV for human health concerns only. Nor is an application required in such cases for the incorporation of the substance into the medical device during the manufacturing process or for the uses and corresponding volumes of that substance upstream preceding the end-use.
In case of any contradictions between the position expressed in this Q&A and the positions expressed previously by ECHA in the RCOM of 20 December 2011, the position expressed in this Q&A should be considered as the current understanding of the law. It takes precedence over any other views communicated previously by ECHA on this issue.
The manufacture of a substance is not subject to the authorisation requirement. After a substance has been manufactured it may have to be handled before it is exported or placed on the EU market. Operations which are necessary for the handling of a substance on its own in the manufacturing for export or placing on the EU market are considered to be part of the manufacturing stage (e.g. filling into appropriate containers, storage, addition of stabiliser, dilution to a safer concentration -if necessary for transport safety-), but not other uses such as the formulation of a mixture or incorporation of the substance into articles. The formulation of a mixture or incorporation of the substance into articles are considered "uses" within the meaning of Title VII of REACH and are subject to the authorisation requirement whether or not the mixture or articles will be exported or placed on the EU market.
No. When a sample containing an Annex XIV substance is taken from a production line for further analysis the sampling activity shall be described and assessed e.g. in a worker contributing scenario that is part of the application for authorisation. However, activities considered to form part of the use of the sample in performing analytical activities can benefit from the exemption under Article 56(3) of REACH. See also Q&A 585.
Yes. The use of an Annex XIV substance when it is required, on its own or in a mixture, as part of an in vitro diagnostic (IVD) method (e.g. in a reagent, calibrator, control material or kit) is considered as scientific research and development and is therefore exempted from authorisation requirements if this activity is carried out under controlled conditions and in a volume not exceeding one tonne per year per legal entity (see Q&A 585).
Annex XIV substances may be required to provide a range of functions during IVD analysis, including: to remove impurities, to prevent undesired reactions that would lead to false positive results, to stabilise or solubilise proteins during analysis, to inactivate viruses prior to analysis.
The exemption covers also IVD medical devices for veterinary and animal health purposes.
It depends. See the more general Q&As 1498 and 1030, as well as Q&A 1442.
For instance, the incorporation of an Annex XIV substance into an IVD medical device which is an article is not exempted under Article 56(3) REACH. Similarly, the use of an Annex XIV substance in upstream life-cycle stages to produce IVD medical devices which are not articles (e.g. reagents, calibrator, control material or kits) and where the Annex XIV substance is not incorporated into the IVD device is not exempted under Article 56(3) REACH. An example of the latter is the use of an Annex XIV substance to lyse cell membranes during the purification of antibodies that are subsequently used in an IVD medical device which is not an article: that use would require an authorisation.
Uses in PPORD are not generically exempted from the authorisation requirement.
However:
- Specific entries in the Authorisation List may include an exemption for uses in PPORD below a specified maximum quantity;
- Activities to develop products and processes may fall under the definition of Scientific Research & Development (SRD) if they are carried out under controlled conditions in a volume less than one tonne per year. In this case, they are exempted from authorisation.
Application for authorisation dossiers shall consist in an IUCLID file, to which a series of specific documents are attached. For the purpose of an application for authorisation, the latest version of IUCLID should be used. Formats for these attachments are provided by ECHA on its website
Authorisation application dossiers shall be submitted electronically via REACH-IT. See (http://echa.europa.eu/applying-for-authorisation)
The whole application can be submitted in any single official EU language. For example, you can submit the whole application in English. However, you cannot submit an application where most of the documents are in English and rest in another language, For details, see question 129.
ECHA would consider those parts of the Application that are not in the main language as "not received". These documents could be essential for conformity of the application under Article 62 of REACH. If so, the Committees would request the applicant to submit an update of these documents in the main language. If the applicant fails to submit the translations, the Committees would not consider the application to be in conformity with the requirements of Article 62.
The Broad Information on Uses (BIU) of the Annex XIV substance refers to a "brief wording" containing:
- information based on the name of the use applied for,
- the use descriptors and function, and
- key information on the conditions of use.
In addition, the BIU contains the following documents:
- the public version of the Analysis of alternatives,
- the public version of the Substitution Plan, if provided in the application,
- the public version of the Socio Economic Analysis, if provided in the application, and
- the public version of sections 9 ("Exposure assessment") and 10 ("Risk characterisation") of the Chemical Safety Report (CSR) covering the use applied for.
For further details and example, see: http://echa.europa.eu/documents/10162/13555/public_information_afa_en.pdf
Yes. The applicant's name will normally be made public as part of the Broad information Use during the public consultation on alternatives.
Include the complete versions of sections 9 and 10 in your Chemical Safety Report (CSR) and attach the CSR to section 13 of IUCLID (naming it for instance "CSR.pdf").
Attach a public version of sections 9 and 10 naming it clearly (e.g. "Public_version_of_CSR sec9-10.pdf") and attach it as a separate document in section 13 of IUCLID.
If all the information contained in sections 9 and 10 can be made public, then these two sections will be published as they are provided in the CSR.
If you (as the applicant) have already registered the Annex XIV substance, the CSR prepared for registration purposes should be the basis for the preparation of the CSR needed in an application for authorisation. However, it is likely that it needs to be updated and adapted to the authorisation context (e.g. regarding the refinement of uses, emissions and exposures, the overall quality of the CSR, etc). When submitting your application you can attach a version of your CSR specifically developed for authorisation. You can use this opportunity in cases, where you may wish to provide – for the authorisation purposes – only an extract of the latest updated CSR submitted in the registration process. This updated CSR would contain only those parts that are relevant for the authorisation application, but developed in more detail.
Or you can simply refer to the CSR provided in your registration dossier. In the latter case you should – if necessary – update your CSR before you submit your application to ECHA. If you develop a specific CSR for authorisation, you should be aware that as a rule the information contained in the CSR submitted in an authorisation application should be in the CSR provided in the registration dossier.
After the sunset date the applicant will need to update the CSR submitted in the registration in order to remove all uses for which he has not applied for authorisation.
Following the authorisation decision taken by the Commission, the Applicant may also need to further update the CSR submitted in his registration.
Applicants can provide information which should not be made public in the "complete versions" of the assessment reports (Chemical Safety Report, Analysis of Alternatives, Substitution Plan, Socio-Economic Analysis). Formats and instructions are available under the "Preparing Applications for authorisation" page (http://echa.europa.eu/applying-for-authorisation/preparing-applications-for-authorisation).
Applicants need to make "public versions" of these "complete versions". In "public versions" the applicants need to blank out confidential business information. The "public versions" will be published on ECHA's website during public consultation on alternatives. Applicants need to provide solid justifications as to why the information which has been blanked out should not be made public.
A joint application for authorisation in REACH is made and submitted simultaneously by a group of applicants (i.e. the submitting applicant and the co-applicants). The submitting applicant starts by uploading the IUCLID dossier in REACH-IT. When the dossier has been successfully uploaded, the submitting applicant receives the joint application name and security token, which he will communicate to the co-applicants.
The submitting applicant and co-applicants indicate the substance-use combinations they are applying for, scope of their use(s) and provide the information on their role in the supply chain by completing the information in REACH-IT. The submitting applicant finalises the joint application after all co-applicants have completed their parts of the information and confirmed their participation.
Due to the possible complexity and technical issues of joint applications, ECHA recommends to prepare and submit a joint application when (i) all co-applicants of the group apply for all uses in the joint application, and (ii) the co-applicants have agreed on a way to share all the information provided in the application.
In complex cases, it may be preferable for each applicant to submit their own application separately. The applicants can cooperate during the preparation of their applications. For details, see the manual ‘How to prepare an application for authorisation manual’.https://echa.europa.eu/manuals, Application for Authorisation Guidance (Appendix 2) and the presentations "Description of uses for authorisation / Broad Information on Uses" and "Joint versus Individual applications" for additional information.
No. In contrast with Joint Registrations, an additional legal entity cannot join the group of applicants which have already submitted a joint application for authorisation.
An applicant may submit a subsequent application in which he can refer to appropriate parts of an application previously submitted for a use of a substance, provided that the subsequent applicant has permission from the previous applicant(s) to refer to these parts. Furthermore, the subsequent applicant shall update the information taken over from the original application as necessary. This subsequent application will be processed and evaluated on its own merits.
Via REACH-IT and the secured webforms. However, for organisational and practical issues, emails can be used.
Yes, if they form a group. The possibility to cover more than one substance in the same application is limited to substances that meet the definition of a group of substances as stated in Section 1(5) of Annex XI of the REACH Regulation. In all other cases, you have to submit a separate application for each substance. See Appendix I of the Guidance on the preparation of an application for authorisation on substance grouping.
When reading the Committees' draft opinions you may have noticed misinterpretations, misunderstandings or even errors that need to be addressed in the draft opinions. ECHA encourages you to provide all the necessary clarifications with the relevant supporting evidence during the commenting period. However, the Committees will only take into account new data or information (e.g. new measurements, figures) that are related to such misinterpretations, misunderstandings or errors noted by the applicant. It is the obligation of the applicant to provide the necessary relevant data at the time of the initial submission of the application or in response to earlier requests from the Committees.
Several scenarios can be envisaged: A base fee is payable, which covers one substance and one use. An additional fee applies for each additional use and substance. No additional fee for additional applicant(s) is levied, though. However, the levels of the base fee and of the additional fees per use and substance depend on the size of the largest company that is party to the application - .i.e. if in joint applications companies are of a different size, the highest applicable fee will be levied.
For example, in the case of an application submitted by four parties with ten three uses and two substances in total, the relevant fee will be the base fee applicable to the largest applicant + three additional applicant fees + nine two additional use fees applicable to the largest applicant + one additional substance fee applicable to the largest applicant.
The table below shows four examples of calculated fee for applications for a single substance where the (largest) applicant is a large company.
Table: Fees depend on the number of uses, not applicants
| Examples | A | B | C | D |
|---|---|---|---|---|
| Applicants | 1 | 2 | 1 | 2 |
| Uses | 1 | 1 | 2 | 2 |
| Fee | €54 100 | €54 100 | €102 790 | €102 790 |
The possibility to cover more than one substance in the same application is limited to substances that meet the definition of a group of substances as defined in Section 1(5) of Annex XI of the REACH Regulation. In all other cases, a separate application must be submitted for each substance.
Further information: ECHA Fee Calculator: a tool provided by ECHA to estimate the possible amount of a fee related to a given application for authorisation.
If you submit an application for an authorisation on your own, you will have to pay a base fee. That fee covers one use and one substance. If you need to apply for authorisation for more uses, you will need to add an additional fee for each additional use you want to cover. Thus, if your application covers three uses, the fee that you will have to pay is the sum of the basic fee + two additional use fees. The level of the base fee and the additional fees depend on the size of your company; reduced fees apply if you are a micro, small, or medium-sized enterprise.
ECHA will calculate the fee based on the Fee Regulation. It has made available a Fee Calculator to help the applicants to know in advance the amount that they would expect to pay.
ECHA will send the invoice always to the legal person that submitted the application. Thus, in a joint application ECHA will send the invoice to that applicant that submitted the application. It is then up to this applicant to split the invoice according to the agreement that the applicants have made between themselves. For clarity, ECHA is not in a position to send separate invoices to each of the parties covered by the application.
ECHA sends the applicant an invoice in about 8 weeks after the submission.
The applicant has 14 calendar days to pay the invoice with a possibility to extend it by another 7 calendar days. Thus, all in all the applicant is given 21 days to pay the invoice from the date it received the invoice.
If the full payment is not made by the extended payment due date, the application for authorisation will be considered as not received by ECHA. In this case the application is not processed further. The only way to proceed is to re-submit the application.
The necessity to impose an additional fee will reflect ECHA's workload associated with processing and evaluating the application. The calculation of the base fee and any applicable additional fees is described in Q&A 600. An application would lead to additional workload if the number of ‘uses applied for' and/or exposure scenarios is greater than one. One "use applied for" shall be covered by at least one exposure scenario. However, the number of uses vs the number of exposure scenarios might not be identical. Thus, in order to reflect the additional workload, ECHA calculates the fee based on the highest number of uses vs. exposure scenarios.
Example 1: Iif you apply for two uses that are covered together in your Chemical Safety Report by one generic exposure scenario, ECHA will calculate the invoice based on two uses.
Example 2: If you apply for two uses which are covered by three different exposure scenarios, ECHA will estimate whether the evaluation of the additional exposure scenario increases ECHA's workload and whether the fee can be based on three uses.
No.
No. Currently ECHA will not impose a fee for an exposure scenario covering an article service life. ECHA needs to gain experience with the first applications for authorisations. ECHA reserves its right to re-evaluate this approach if, based on the experience gained with handling this type of exposure scenarios in the context of an Applications for Authorisation, ECHA concludes that the associated workload warrants a fee. It is also possible that a fee may be imposed for instance, in conjunction with a review of the Fee Regulation in the future.
Teleconference based information sessions (TIS) give an opportunity for future applicants to clarify regulatory and procedural aspects of the authorisation application process, and for ECHA to have a better understanding of the practical issues related to Applications for authorisation. ECHA organised earlier “Pre-submission information sessions” (PSIS). TIS are a more focussed, teleconference based way of fulfilling the same purpose as with PSIS.
For further details see ECHA's Support webpage on teleconference based information sessions (link to: https://echa.europa.eu/applying-for-authorisation/pre-submission-information-sessions).
You can make a request for a teleconference based information session (TIS) either when notifying ECHA of your intention to submit an application for authorisation, or later. You should nevertheless send your request for a TIS at least eight months before the planned date for the submission of your application for authorisation. Although you can still notify less than eight months in advance of the submission of your application, this may have an impact on ECHA's availability to organise a TIS for your company. Please bear in mind the TIS should take place within one month after the meeting request and that you probably would like to have sufficient time after the TIS to potentially further work on your application before submitting it to ECHA.
Requests for TIS are made via a secure web form, available on ECHA's website. For further details see ECHA's Support webpage on TIS (link to: https://echa.europa.eu/applying-for-authorisation/pre-submission-information-sessions).
A CSR has to be submitted as part of the application. This CSR can be (i) the applicant's own CSR for authorisation, or (ii) a reference to CSR submitted by the same applicant for registration, or (iii) a reference to CSR of a previous applicant (case of subsequent applications for authorisation). If the applicant for authorisation is a Downstream User (i.e. not a registrant) and he generates his own CSR for the authorisation, this CSR can be based on updated information from the registrant's CSR as long as he has the right to use the information from the respective data owners.
However, data sharing obligations as specified under Art 30 of REACH do not apply for authorisation. The reproduction or further distribution of information from Registration Dossiers and Notifications to the C&L Inventory which are published on ECHA's dissemination website may be subject to copyright protection. Furthermore, the use of the information without obtaining the permission from the owner(s) of the respective information might violate the rights of the owner. The Agency does not take responsibility for any copyright or other infringements that may be caused by using the information.
Potential applicants are reminded that they shall ensure that they have adequate rights to submit this information to the Agency and acknowledge that this information can be used for the purpose of the application for authorisation.
See also question 916
In joint applications, only one PSIS will be available, and this session is for the group of applicants as a whole. Due to the limited availability of places for the PSIS, it is for the group of applicants to decide who would be the most appropriate to represent them in the session.
The description of the ‘uses applied for' and of the related exposure scenarios is key step in the preparation of an application for authorisation. The concept of "use applied for" in the context of applications for authorisation might significantly differ from what industrial actors usually understand by a "use" in their day-to-day practice. The ‘use applied for' description, scope and level of details are not only related to the risks linked to the exposure/release of the Annex XIV substance but also to the analysis of alternatives and, where relevant, to the socio-economic analysis and substitution plan. The following documents describe this issue in more details:
- How to develop the description of uses in the context of Authorisation [PDF] [EN]
- Presentations from seminars on applications for authorisation
- Guidance on information requirements and chemical safety assessment - Exposure Scenario Format
The demonstration of adequate control or minimisation of the risks should be made in the Chemical Safety Report (CSR) of the application for authorisation. REACH Annex I defines the Derived No-Effect Level (DNEL), i.e. the level of exposure above which humans should not be exposed and the way the risk characterisation for the human population should be performed, based on the DNEL (if a DNEL can be determined). DNELs derived according to the ECHA guidance are therefore the reference data for the demonstration of adequate control.
It should be noted that the Committee for Risk Assessment (RAC) has derived reference DNELs for some substances on Annex XIV and intends to continue doing so for other Annex XIV substances.
For more details, see:
An application for authorisation is not a matter of pages but rather how convincing the argumentation is. All the necessary information for the ECHA Committees for making their opinions and for the Commission to make its decision should be included in the application in a clear and convincing way. Overall in a well-argued application, strong or clear messages should not be diluted or obscured by lengthy text. Also you can consider using appendixes to provide additional, complementary information.
Yes, as long as the application is in conformity with the rest of Article 62 and the applicant decides to use in his assessment the DNELs or dose response relationships derived by RAC for the intrinsic properties of the substance specified in Annex XIV. In this case the applicant does not need to provide the hazard data necessary to derive DNELs or dose response relationships.
Yes, the Chemical Safety Report shall always include the "Part A" duly completed. Part A contains the Summary of Risk Management Measures and the declarations that these measures are implemented and communicated. See also Q&A 611.
Usually not. A suitable alternative should results in an overall reduction of risks to human health and the environment compared to the Annex XIV substance. An alternative substance having a similar (eco)toxicological profile to the Annex XIV substance (for instance a SVHC substance) is unlikely to meet the overall reduction of risks criterion. However, even if the alternative does not lead to an overall reduction of risk, this alternative substance should still be part of your analysis of alternatives but the conclusion regarding the overall reduction of risks should be relatively straightforward. As a consequence, you may consider whether a detailed analysis of its technical and economic feasibility is necessary in such a case.
As a result of the eight-week public consultation both the "complete" and the "public" versions of the comments on the alternatives are made available to the Rapporteurs and the Committee members. The Rapporteurs will use this information as any other information during an evaluation process. The relevance, validity and reliability of the information will be assessed. The applicant will have a possibility to respond to the "public versions" of the comments. The Rapporteurs may pose follow-up questions to the third parties. They may also pose questions or require additional information from the applicant, as a result of the information provided by third parties.
Both the applicant's and society's perspectives are relevant in the application. The applicant's perspective is relevant in the Analysis of Alternatives to assess how feasible it would be for the applicant to adopt any alternatives which are available to him. This would include the alternative of ceasing use of the substance altogether. This analysis can be used to identify the non-use scenario(s) which would pertain, if authorisation is not granted. The use and non-use scenarios should be assessed from society's perspective to provide an assessment of whether authorisation is justified from a social perspective or not, based on a comparison of the benefits of authorisation and the risks.
The applicant should undertake an Analysis of Alternatives as if they will be unable to use the substance in question beyond the sunset date, that is, as if their application for authorisation is not granted. Thus, the scope should be whatever the applicant would normally use when choosing alternative substances or technologies in his business. One qualification to this is that alternatives might be suggested through the public consultation of which the applicant was previously unaware or would not normally consider. He might wish, therefore, to extend the normal scope of his analysis in anticipation of this.
It is not possible to generalise about the exact scope of the socio-economic analysis, as this will vary on a case-by-case basis. The principle is that all factors should be incorporated to the extent that they might make a material difference to the conclusions. An iterative approach to developing the analysis, as well as consultation and input from socio-economic analysis experts, can be helpful in establishing the appropriate scope in any given situation.
More details in the presentations of the seminars on application for authorisation:
A Substitution Plan should be provided if suitable alternatives are available (cfr. Article 62(4f)). A Substitution Plan is a commitment to take the actions needed to substitute the Annex XIV substance with a suitable alternative substance or technology within a specified timetable. If you conclude that the alternatives you have identified are neither suitable nor available yet, you should not submit a separate Substitution Plan. You should rather include all the relevant information on R&D activities in your Analysis of Alternatives. Refer to the Guidance on Application for Authorisation for more information.
There is no requirement to submit a socio-economic analysis when submitting an application for authorisation under the adequate control route. However, ECHA recommends that a targeted socio-economic analysis is also prepared under the adequate control route. This would be helpful in particular to the Committees when proposing to the Commission the duration of the review period. Furthermore, if the ECHA Committees do not support the applicant's assessment according to which the risks are adequately controlled, the application can still be evaluated on the provided socio-economic analysis.
Based on Articles 60(4) and 64(4)(b) of the REACH Regulation the Committee for Socio-economic Analysis (SEAC) needs to state in its opinion, if the alternatives to the substance that is applied for are suitable. Economic feasibility is one aspect of this. It is recognised that the evaluation of economic feasibility is intrinsically linked with the evaluation of the other aspect of suitability of the alternatives, i.e. technical feasibility. Usually the more difficult (or easy) it is to substitute a substance the more expensive (or cheap) it is to do this. The note below describes how SEAC intends to evaluate economic feasibility as part of applications for authorisation recognising the link with technical feasibility.
The applicant should undertake an Analysis of Alternatives as if they will be unable to use the substance in question beyond the sunset date, that is, as if their application for authorisation is not granted. Thus, the level of detail should be whatever the applicant would normally use when choosing alternative substances or technologies in his business. However, as stated in the guidance on applications for authorisation: "It is strongly recommended that the applicant demonstrates that a comprehensive and adequate assessment of alternatives has been done. This is because the Agency in its opinions and the Commission in its assessment of whether suitable alternatives are available will take "all relevant aspects" into account {Art. 60(5)}, including information submitted by interested third parties." However, regarding the assessment of the risks arising from the use of the alternative and the comparison of these risks with the ones arising from the use of the Annex XIV substance "the applicant is not required to generate new hazard data or provide a chemical safety assessment for each of the alternatives. Nor is it required that the risks associated with alternative substances or technologies are assessed in the same detail as the risks associated with the Annex XIV substance."
In conclusion, the level of effort that needs to be put into the analysis of alternatives will be a matter of judgment for the applicant and should reach a point where the applicant is comfortable in defending its analysis in front of the ECHA Committees.
Yes, if they are relevant. However, the focus of the socio-economic analysis should be the EU but information on effects outside the EU may be relevant as well. See chapter 2.4.3 in the "Guidance on the preparation of socio-economic analysis as part of an application for authorisation"[PDF] for more information.
Sometimes an applicant may not have been profitable in recent years but still seeks an authorisation for continuing the use of a SVHC. The question thus arises regarding how not obtaining an authorisation would affect the applicant. To answer this question, it should first be noted that changes in applicants’ profits are only one possible element to consider when estimating the benefits of continued use. Impacts on other actors (e.g. within the supply chain, on alternative providers or consumers) may also be important. Other approaches, such as the compliance cost guidance, may also be relevant when assessing the benefits of continued use.
When considering changes to profits as a measure of benefits of continued use, the assumptions about future profits in the baseline scenario need to be justified, regardless of whether the company in question has had negative profits in the past or not. If the company has had negative profits, it is also useful to explain the reasons for this. In particular, the applicant should clarify whether these negative profits link directly to activities that rely on the use of the SVHC. If there is a specific reason why the company has had negative profits in the past, but this is not expected to be the case in the future, then it may make sense to base future projections on, for example, the profits of a typical year or period. The profits considered should be those related to the use applied for (and not e.g. other products of the company not related to the use of the SVHC).
Once the total expected profits over the baseline scenario have been established (with any expected negative profits in specific years reducing the total profits made), an approximative yearly profit can be calculated by dividing the total profits with the number of years applied for. More accurately, the applicant can derive an annuity of expected profits by using the standard annuity formula PMT(rate, nper, pv, [fv], [type]) in Excel.
The Annex XIV entry for HBCDD is a relatively broad entry, which might be considered to describe UVCB substances (with undefined Br positions), as well as multi-constituent substances (containing more than one defined diastereoisomers at concentrations between ≥10% and <80%), and mono-constituent substances (one diastereoisomer ≥80%).
If the application concerns one particular HBCDD substance (either UVCB, or multi- or mono-constituent), then sections 1.1 and 1.2 should describe this specific substance, following the general instructions provided in the Manual: How to prepare an application for authorisation (link to: https://echa.europa.eu/manuals).
Should the application concern more than one substances covered by the HBCDD entry (for instance due to different manufacture processes by one manufacturer, or in case of a joint application with applicants manufacturing/importing significantly different HBCDD substances), the application can still be considered as an application for "one Annex XIV substance". Therefore ECHA will charge a fee for "one substance", and the IUCLID dossier shall accordingly contain one substance dataset.
In this latter case, for technical reasons in section 1.1 of the IUCLID application dossier the following information should be filled in: IUPAC name: Hexabromocyclododecane; Type of substance – Composition: other: multiple substances under Annex XIV entry. The actual compositions of the substances applying for can be listed individually as different compositions in section 1.2 by using the repeatable block-function. In the "Brief description" field of each listed composition, please indicate also a remark, e.g. "substance manufactured from manufacturing site A" or "substance manufactured/imported by applicant B".
It is reminded that the REACH Annex XIV substance entry number (for HBCDD this number is "3") shall always be given in section 1.3 of the IUCLID dossier.
General instructions on how to prepare an IUCLID application for authorisation dossier are provided in the Manual: How to prepare an application for authorisation (link to: https://echa.europa.eu/manuals)
Yes. The reasoning and some example are provided below.
When brought in contact with water, chromium trioxide (EC number 215-607-8) forms two acids and several oligomers: Chromic acid (EC number 231-801-5), Dichromic acid (EC number 236-881-5), Oligomers of chromic acid and dichromic acid (further referred as "Chromic acids and their oligomers"). These chemical species are all identified as substances of very high concern (SVHC) and included in Annex XIV1 as two separate entries. Latest Application Date (21 March 2016) and Sunset Date (21 September 2017) are identical for both entries.
Chromic acids and their oligomers generated in water from chromium trioxide are commonly referred to as an aqueous solution of chromium trioxide. With regard to the authorisation requirements, it may be justifiable in some situations to consider for practical reasons chromic acids and their oligomers as an aqueous solution of chromium trioxide.
The generation of chromic acids and their oligomers by adding chromium trioxide to water is a use of a substance under REACH. This may be a discreet use of chromium oxide by a formulator or part of an integrated process in the use of chromium oxide. Considering this in the context of applications for authorisation, this operation should be considered as a "use applied for" and be addressed in the chemical safety report (CSR), the analysis of alternatives and, if appropriate, in the socio-economic analysis. Hence, applications for authorisation that are meant to cover further uses down the supply chain after the initial step in which chromium trioxide is brought in contact with water, have to refer to the chromic acids and their oligomers.
This answer is strictly limited to chromium trioxide and chromic acids and their oligomers generated from chromium trioxide in water. The system in aqueous solution is a complex equilibrium between multiple chemical species which depends on several physico-chemical parameters and the different chemical species cannot be isolated from the aqueous solution. The answer should thus not be applied by analogy to any other substance.
The table below describes possible scenarios in the case of an application for authorisation is made by the manufacturer/importer/user of chromium trioxide and/or chromic acids and their oligomers. Technical instructions are provided in the last column of the table.
As a general rule, sections 1.1, 1.2 and 1.3 of IUCLID should always refer to the substance applied for (i.e. the substance indicated in the third column - "Application for authorisation" of Table 1). In addition, if there is more than one substance which is imported/used (scenario #2 below) or if the substance actually imported/used is different from the substance applied for (scenario #3b below) this information should be described in section 1.2.
For each use, the assessment performed in the assessment reports (CSR, analysis of alternatives (AoA) and socio-economic analysis (SEA)) should relate to the substance relevant for that use. The substance at the use level (e.g. chromic acids and their oligomers - used in electroplating) might indeed differ from the substance applied for (e.g. chromium trioxide) in terms of hazard, physical form / potential for exposure to Cr(VI), alternatives, etc.
Table 1 - Possible scenarios in the case of an application made by the manufacturer/importer of chromium trioxide and/or chromic acids and their oligomers2
| Scenario # | Actor / Scenario | Application for authorisation | Explanation / Technical instructions |
|---|---|---|---|
| 1 | Manufacturer / importer/User of chromium trioxide who generates chromic acids and their oligomers in water applies for an authorisation for this use and the further uses of chromic acids and their oligomers (by itself and/or its downstream users) | One application for authorisation for chromium trioxide covering the further uses of the chromic acids and their oligomers |
Substance ID in IUCLID
|
| 2 |
Importer/User of both
applies for an authorisation for the generation in water of chromic acids and their oligomers and their further uses (by itself and/or its downstream users) |
One application for authorisation for chromium trioxide covering the further uses of the chromic acids and their oligomers |
The application should be made for chromium trioxide making clear that chromic acids and their oligomers are also imported/used. Substance ID in IUCLID:
Assessment reports in IUCLID (CSR, AoA, SEA):
|
| 3 | Importer/User of chromic acids and their oligomers generated in water from chromium trioxide applies for an authorisation for the further uses of chromic acids and their oligomers (by itself and/or its downstream users) |
One application for authorisation either for
|
In case the importer/user decides to apply for an authorisation for chromic acids and their oligomers in an application for chromium trioxide (case b), it has to become clear from the application that what is actually imported/used are chromic acids and their oligomers: Substance ID in IUCLID:
Assessment reports in IUCLID (CSR, AoA, SEA):
|
| 4 |
Manufacturer/Importer/User of chromic acids and their oligomers generated by alternative methods other than from adding chromium trioxide to water3 or Importer/User who is unaware of the manufacturing methods of the chromic acids and their oligomers applies for an authorisation for the further uses of chromic acids and their oligomers (by itself and/or its downstream users) |
One application for authorisation for chromic acids and their oligomers covering the further uses |
The approaches described above cannot be applied as the starting material for manufacturing chromic acids and their oligomers is not chromium trioxide or is not known. Substance ID in IUCLID:
Assessment reports in IUCLID (CSR, AoA, SEA):
|
Please see also Q&A=804 (Do chromic acids and their oligomers, generated in water from chromium trioxide, require their own registration under REACH?)
1 Entry#17: 'Acids generated from chromium trioxide and their oligomers. Group containing: chromic acid, dichromic acid, and oligomers of chromic acid and dichromic acid' And Entry#16: 'chromium trioxide'
2 Chromic acids and their oligomers generated from chromium trioxide could be described in IUCLID sections 1.1 and 1.2 e.g. as a UVCB substance with IUPAC name "Acids generated from chromium trioxide and their oligomers" and Brief description "complex composition typically including oligomers of chromic and dichromic acids in equilibrium with each other". As constituents there could be listed i) chromic acid, ii) dichromic acid, and iii) oligomers of chromic and dichromic acids.
3 Further clarification may be needed as to whether alternative methods would lead to similar equilibrium as for aqueous solutions
These Annex XIV entries are relatively broad entries, covering well-defined substances and UVCB substances, polymers and homologues.
If the application concerns one particular substance within the scope of one of these entries, then sections 1.1 and 1.2 should describe this specific substance, following the general instructions provided in the Manual: How to prepare an application for authorisation.
If the application concerns more than one substance covered by the applicable entry (i.e., either entry 42 or entry 43 in the authorisation list), the application will still be considered as an application for "one Annex XIV substance" for that specific entry. Therefore ECHA will charge a fee for "one substance", and the IUCLID dossier shall accordingly contain one substance dataset.
In this latter case, for technical reasons section 1.1 of the IUCLID application dossier should contain the following information:
- IUPAC name: the name of the entry as it appears in the Authorisation List (“4-Nonylphenol, branched and linear” or “4-(1,1,3,3-tetramethylbutyl)phenol, ethoxylated” accordingly);
- Type of substance – Composition: “other:” “multiple substances under Annex XIV entry”.
In this case, in section 1.2 the available information about the actual compositions of the substances applying for can be provided, listed as different compositions by using the repeatable block-function.
It is reminded that in all cases the REACH Annex XIV substance entry number (for these entries the number is "42" and “43” accordingly) shall be given in section 1.3 of the IUCLID dossier.
General instructions on how to prepare an IUCLID application for authorisation dossier are provided in the Manual: How to prepare an application for authorisation.
It is noted that in the case the application concerns both entries (42 and 43) a single multi-substance dossier will need to be submitted. In this case, ECHA will charge a fee for two substances and the IUCLID dossier shall accordingly contain two substance datasets, according to the instructions provided in the manual.
Yes, given the method of manufacture of this type of substance. The commercially manufactured nonylphenol ethoxylates predominantly consist of C9 alkyl substituents in position 4 (para-) of the phenol ring. Therefore a substance defined as nonylphenol ethoxylated is covered by the entry 43 for ‘4-Nonylphenol, branched and linear, ethoxylated’.
A change of legal entity is a change of the legal person who is the applicant of an authorisation or an authorisation holder. This change can take place, for instance, as the result of a merger, a split or an asset sale (sale of a production site or business). See Q&A 1242. Also purely administrative changes such as a change of corporate name or address and a change of Only Representative (OR) of a non-EU company need to be notified. See Q&A 1253.
Yes.
Please submit the notification as follows:
- Go to REACH-IT and use the “change name” functionality to update the name. Please provide in REACH-IT an extract from commercial register as evidence of the change.
- Then notify ECHA via email: legal-entity-changes-authorisation@echa.europa.eu. Please mention the affected submission number (of application) or authorisation number (of granted authorisation), and confirm that you have updated the name in REACH-IT.
In this manner ECHA can quickly process the change. ECHA will verify the evidence of the change and inform you as soon as it has updated its databases. In case of a granted authorisation, ECHA will also forward your notification to the European Commission.
Yes. An application for authorisation or a granted authorisation can be transferred as long as the transfer is the result of the change of legal entity referred to in Q&A 1239 and the person to whom it is transferred qualifies as manufacturer, importer or downstream user with regard to the substance(s) and the use(s) covered by the application for authorisation or the decision. For details, see Q&As 1239 and 1242. On Only Representatives, see Q&A 1250.
Please note that a change of legal entity cannot extend the scope of the original application for authorisation or of the authorisation, e.g. to cover different uses.
Please follow the next steps:
- If relevant, create a new REACH-IT account for the successor legal entity. This is relevant when the new legal entity did not have a REACH-IT account before.
- Log into the REACH-IT account of the original legal entity and from the Menu select Manage company | Legal entity change | Initiate a legal entity change. Then follow the online instructions. For the submission, include two attachments:
- Evidence of change, for instance the merger agreement or the agreement of the sale of assets.
- Filled announcement using the format indicated below. This describes the change of legal entity and the concerned application(s) or authorisation(s), as well as provides an analysis of the key impacts. It needs to be signed by both the original and the successor legal entity.
See also Q&A 1247.
Remember, that if the original entity has other REACH assets (e.g. registrations), the change of legal entity may need to be notified, too, via the same functionality in REACH-IT. See ECHA’s Practical Guide and technical instructions provided in the REACH-IT tool.
- Format [DOCX]
Currently no fee needs to be paid for this notification for the purposes of authorisation.
If the change concerns a pending application for authorisation, ECHA will communicate whether the notified change corresponds to the requirements described in Q&A 1241 and is properly documented. Furthermore, it will give its view on whether the change is minor (i.e. purely administrative changes and changes that would not have material implications on the content of the application or the terms of the RAC and SEAC opinions) or major (i.e. all other cases). ECHA endeavours to process notifications in two weeks. If the change is confirmed, ECHA will provide to the original legal entity a token. The original legal entity will need to communicate the token to the successor legal entity, who will need to accept the legal entity change in REACH-IT. The change is considered valid as of the date of the notification.
In this case ECHA will amend its database concerning the change of the legal entity. If the change is major, the RAC and the SEAC might need additional time to assess the implications of the change during the opinion-making process. If ECHA has already adopted an opinion, it will send its assessment to the European Commission so that it can take this into account when deciding on the authorisation.
In cases concerning granted authorisations, ECHA will send its assessment on the nature of the change to the European Commission, including its assessment on whether a review of the authorisation should be triggered on the basis of Article 61(2) of REACH.
The timelines for the assessment about any implications for the RAC and SEAC opinions or for the authorisation decision (potential trigger of a review of the authorisation) depend on the stage in the process at which the notification was made.
ECHA or the Commission may request for clarifications or additional information concerning your notification.
An applicant for authorisation or an authorisation holder should inform ECHA about any legal entity change as soon as it has taken place (i.e. when they have the documentary evidence) using the ‘Legal entity change’ functionality in REACH IT. To reduce uncertainty you can also consult ECHA on the specific case in advance, provided you have sufficient information about the foreseen change. For any questions, please contact ECHA at legal-entity-changes-authorisation@echa.europa.eu.
Yes.
Please proceed as follows:
- Create a new REACH-IT account for the new Only Representative.
- Log into the REACH-IT account of the original Only Representative and from the Menu select Manage company | Legal entity change | Initiate a legal entity change. Then follow the online instructions.
ECHA will verify the evidence of the change and inform you as soon as it has updated its databases. In case of a granted authorisation, ECHA will also forward your notification to the European Commission.
Yes.
Please submit the notification as follows:
- Log into REACH-IT and use the “change name” functionality to update the name. Please provide in REACH-IT an extract from commercial register as evidence of the change.
ECHA will verify the evidence of the change and inform you as soon as it has updated its databases. In case of a granted authorisation, ECHA will also forward your notification to the European Commission.
The Review Report updates the Application for Authorisation (AfA), and it must be sent to ECHA. The fee is the same as the one for an AfA. The Review Report will follow the same process as for an AfA. The greater the relevant technical and other progress during the review period, the greater the need to update the information. The information elements are the same. In other words, a Review Report should include an updated version of the assessment reports submitted in the original application (Chemical Safety Report, Analysis of Alternatives and – when relevant – an updated Socio-economic Analysis and an updated Substitution Plan). The use description may also need to be updated e.g. to reflect a more narrow scope.
See Section 6 of the Practical Guide “How to apply for an authorisation” at https://echa.europa.eu/documents/10162/13637/apply_for_authorisation_en.pdf/bd1c2842-4c90-7a1a-3e48-f5eaf3954676.
All the Q&As related to Applications for Authorisation are also valid for Review Reports mutatis mutandis.
You will need to first establish if the increase in the tonnage for the authorised use of the Annex XIV substance would result in an increase in exposure levels to workers and/or emissions to the environment compared to the levels set out in the CSR referred to in the authorisation decision. This CSR forms part of the conditions under which the authorisation was granted to you and served as a basis for the European Commission’s conclusion that the conditions for granting the authorisation were fulfilled.
If you conclude that the exposure levels are likely to increase, you should submit a review report earlier than what was stipulated in the authorisation decision, especially if the increase in exposure leads to higher health and environmental impacts than when the use of the Annex XIV substance was authorised.
You can update your downstream user notification of authorised uses via the relevant reference number page.
Step 1: In REACH-IT, search for the relevant “reference number”
Step 2: Enter an identifier and choose “Downstream user notification of authorised uses” as a dossier type. Click on “Search”
Step 3: The relevant result will be displayed under “Results” page. Click on the reference number.
Step 4: On the reference number page, click on “Create and submit online an update”

Step 5: To continue to the online dossier creation, click on “Yes I want to prepare an online dossier”

Step 6: You are redirected to the online dossier creation. Add any relevant information.
Step 7: In “Update details”, add a reason for updating your downstream user notification of authorised uses.
Carefully review all the information before submitting your dossier.
You can cease all notified uses from the reference number page. Below are the steps on how to cease manufacture in REACH-IT.
Step 1: In REACH-IT, search for the relevant “reference number”.
Step 2: Enter an identifier and choose “Downstream user notification of authorised uses” as a dossier type. Click on “Search”.

Step 3: All downstream user notifications of authorised uses will be displayed. Click on the reference number for the
substance for which you wish to notify you are ceasing the use.

Step 4: On the reference number page, click on “Cease all notified uses”.

Step 5: Choose a justification for ceasing the use, and click on “Confirm the cease use”.

Step 6: The cease use has been successfully recorded. The reference number status is set as “Inactive”.

You should update your downstream user notification of authorised uses to attach your measurements. Please refer to Q&A 1794 to update your downstream user notification of authorised uses.
Once you have launched the submission wizard, click on ‘Edit Authorised uses’ as shown in the screenshot below:

Upload your document (e.g. filled in template provided by ECHA or by the authorisation holder, laboratory analysis etc.) in the ‘File attachment’ section. Read the text in red and take note of the deadline for providing the relevant attachment.

Registrants cannot challenge the inclusion of a substance into the CoRAP (Community Rolling Action Plan).
However, registrant(s) may provide input into the evaluation process. Registrant(s) of a substance in the CoRAP, particularly those substances on the current year of the CoRAP are encouraged to contact the evaluating Member State Competent Authority (eMSCA) early on in the evaluation process. The contact details for the eMSCA are published in the CoRAP. If the dialogue has not already started, the eMSCA will usually contact the lead registrant when the evaluation starts and offer the opportunity to meet to discuss technical issues related to substance evaluation. The registrants should consider nominating a representative for interacting with the eMSCA. The interaction between registrant(s) and the eMSCA at this phase of the evaluation is informal.
When the outcome of a substance evaluation is that an information request to clarify the suspected concern is deemed necessary (i.e. the evaluating MSCA prepares a draft decision on substance evaluation), the registrant(s) will get the opportunity to formally comment within 30 calendar days on any draft decision as part of the decision making process (Articles 50-52 of the REACH Regulation). The evaluating MSCA shall take the comments of the registrant(s) into account and decide whether the draft decision needs to be amended on the basis of the comments/additional information provided by the registrant(s) (Article 50(1) of REACH).
ECHA communicates to the registrant(s) the draft decision as notified to the other MSCAs and ECHA and the received proposals for amendment. At this stage, the registrant(s) will have the opportunity to comment on the proposals for amendment (Article 51(5) of REACH). Registrants are reminded of their obligation to always keep their registration dossier up to date, cf article 22(1).
Substance Evaluation is an integral part of the REACH implementation. It aims to clarify whether a substance, which has been identified as being of potential concern, poses an actual risk to human health and/or the environment. To clarify the risks, the registrants may be asked for more information on the substance. Substance evaluation shall be carried out by the Member States, whilst ECHA coordinates the procedure. The substances to be evaluated annually are listed in the CoRAP (Community Rolling Action Plan).
Member States may volunteer to evaluate a substance. Two Member States may also decide to make a joint evaluation. However, in all cases only one Member State will be designated as the responsible Member State for the evaluation. Final allocation of the substances to the Member States is decided with the adoption of the CoRAP by ECHA, based on the opinion of the Member State Committee (MSC) on the draft CoRAP. Thus, the CoRAP will include for each substance on the list the Member State responsible for the evaluation. The contact information of the responsible competent authority will also be reported for the substances to be evaluated in the first year, to inform the stakeholders about the body handling each substance.
In case of joint evaluations by two Member States, the co-evaluating Member State is also indicated in the CoRAP.
From the publication of the CoRAP, the respective Member States have one year to evaluate substances listed for the first year (n) and, where regarded as necessary, to prepare a draft decision for requesting further information to clarify the suspected risks. The evaluation of the substances listed for the second and third year starts only after the publication of the CoRAP update in year n+1 and year n+2 respectively.
Dossier evaluation comprises the examination of testing proposals and compliance check of registration dossiers. Testing proposals are triggered by the REACH information requirements and all testing proposals submitted by the registrants must be examined by ECHA. The aim of this examination is to decide on the most appropriate testing in order to fulfil the REACH information requirements. ECHA can perform a compliance check on any registration dossier to verify whether the REACH information requirements are met. Dossiers can be chosen for compliance check based on random selection or prioritised based on a specific concern.
The substance evaluation process is triggered as a result of risk-based concerns and aims to clarify whether a substance poses a risk for human health or the environment. Substance evaluation is targeted at substances (including aggregated tonnages, all uses, etc.). Under substance evaluation any information (beyond the REACH requirements) can be requested provided that it is considered necessary for the purposes of risk assessment of the substance.
Substance evaluation is carried out by the Member States, while ECHA is responsible for dossier evaluation.
The decision making process is essentially the same for both processes.
Substance Evaluation may identify risks that could otherwise be missed. This process can further create additional value in respect of:
- Concerns that go beyond the control of the individual registrant, like regional risks or the potential additional risk caused by aggregated exposures of a (sub)population or releases into the environment.
- The assessment of groups of similar substances to predict cumulative effects and potentially increased risk levels from exposures to the different substances in the group.
- If considered scientifically necessary and proportionate, the request for additional information can go beyond the standard information requirements in REACH.
The REACH Regulation Article 44(1) provides the general criteria for substances to be selected for substance evaluation. The legal text defines that prioritisation shall be on a risk-based approach. According to Article 44(1): "(...) the criteria shall consider:
hazard information, for instance structural similarity of the substance with known substances of concern or with substances which are persistent and liable to bio-accumulate, suggesting that the substance or one or more of its transformation products has properties of concern or is persistent and liable to bio-accumulate;
exposure information; tonnage, including aggregated tonnage from the registrations submitted by several registrants".
The criteria have been refined by ECHA in cooperation with the Member States and are published on ECHA's website: Selection criteria to prioritise substances for Substance Evaluation (2011 CoRAP selection criteria) http://echa.europa.eu/documents/10162/13628/background_doc_criteria_ed_32_2011_en.pdf.
These criteria are applied in the initial step of the identification of substances with potential concerns. A further screening and selection process takes into consideration whether the substances are already subject to regulatory measures and the effectiveness of the substance evaluation to clarify the concern by requesting further information on the substance. Thus, meeting the risk-based criteria alone does not automatically mean an inclusion of the substance in the CoRAP.
According to Article 45(5) of the REACH Regulation, a Member State may notify ECHA of a substance, whenever it is in possession of information suggesting that the substance is a priority for evaluation. Thus, the CoRAP may also contain substances that have been included based on notifications from Member States.
Both hazard and exposure information (or a lack of it) is taken into consideration upon prioritising the substances. In the first CoRAP with many substances, the initial concerns are generally related to potential PBT-properties, suspected endocrine disruption, or carcinogenic, mutagenic and reprotoxic properties in combination with wide dispersive or consumer use(s) and/or high tonnages. In general, the uses of these substances cover various areas and do not focus on any particular industrial, professional or consumer uses.
The final published CoRAP also contains a general indication of the reasons why the substance was prioritised and selected for substance evaluation (grounds for concern). Before inclusion in the CoRAP, the substances have not been evaluated and thus the indicated grounds for concern are just an indication of the possible areas of risk, based on the selection criteria. The initial grounds for concern should not be taken as a statement on a known risk or as a statement on what the evaluation will cover. During the evaluation, other areas of concern may be identified and investigated further. Only after the substance evaluation is completed, risks can be defined and communicated. The Member States have agreed that from the first update of the CoRAP in 2013, more detailed justification documents regarding selection of each substance will be prepared and published.
The listing of a substance on the CoRAP does not in itself have any legal impact on the registrant and thus does not require any further action by the registrants. When the CoRAP is adopted/updated, the registrants of substances listed for the first year of the CoRAP may expect to receive a draft decision requesting further information after the evaluation period of 12 months. At that point of time, the registrants will be given an opportunity to comment before any final decision to request further information is taken. The final decision will contain a deadline by which the additional information must be submitted.
On the other hand, if the evaluating Member State comes to the conclusion that no further information is necessary to clarify the risk, the substance evaluation process is concluded without a decision to request further information.
Inclusion of a substance in the CoRAP does not automatically mean that the substance poses a risk to human health or the environment, but rather that there is a concern that it may pose a risk, which needs to be clarified (confirmed or dismissed). It also does not automatically trigger, for example, the restriction or authorisation process. However, the Member State responsible for the evaluation of a substance may consider these options once the evaluation is finished, if the risk is confirmed.
The REACH Regulation does not foresee any formal interaction during the 12 month evaluation process i.e. before the possible draft decision is prepared. Once the draft decision is issued, the registrants will be contacted via REACH-IT. During the decision making procedure registrants will be consulted on any prepared draft decision and proposals made to amend this draft decision. The registrants may submit comments that will be taken into account in the decision making of the evaluating Member State and by the Member State Committee, if the draft decision is referred to the Committee.
The possibility for registrants/stakeholders to interact with the evaluating Member State during the evaluation phase may differ between Member States and substances that are evaluated. However, interaction between the registrants / stakeholders and the evaluating Member State is appreciated in general under substance evaluation. Thus, all relevant information available to the registrants of the substances should be included in the registration dossiers by the start of evaluation (i.e. March each year).
If the evaluating Member State considers that further information is necessary to clarify a potential risk caused by the substance, it may draft a decision specifying the additional data requests. The initially identified concern in the CoRAP does not limit the evaluation made by the Member States and thus the potential request can address any property or exposure scenario of the substance. The registrants of that substance will have an opportunity to provide comments on the draft decision. Such a draft decision will be reviewed and agreed by the other Member States and ECHA, and in the case of proposals for amendment also by the Member State Committee. After this procedure, ECHA will take the final decision in line with the agreement in the Member State Committee. If no unanimous agreement is reached by the Member State Committee, the decision will be taken by the European Commission. The decision will contain a deadline by which the registrants must submit the requested information. It may also be that no request for information is needed because the risks can be clarified with the information already available. In such cases, the substance evaluation is considered to be completed.
Once the registrants submit the requested information, the responsible Member State has another 12 months to assess this information and decide whether a further request for information is necessary or whether the evaluation can be concluded. In this latter case, the responsible Member State should consider whether and how to use the information obtained for the purposes of Community level risk management measures. The Member States may conclude:
- EU-wide risk management measures are necessary (e.g. EU wide restriction, EU-wide authorisation, EU-harmonised classification and labelling, occupational exposure limits, measures for the protection of the environment under the Water Framework Directive) or
- Actions at national level should be taken.
The conclusion can also be that the risks are sufficiently under control with the measures already in place. ECHA informs the European Commission, the registrant and the other Member States about the conclusions.
The decisions on data requests and evaluation reports will be made publicly available once finalised. It should be noted that as the production of the information requested may, in some cases, take several years (e.g. in the case of long term studies and annual environmental monitoring) finishing a final evaluation report may also take several years from the adoption of the CoRAP.
From the publication of the CoRAP (March each year), the Member States have 12 months to prepare a draft decision for a substance included for evaluation during the first year, i.e. by end of February of the following year. After that, the decision making process may take approximately four to eight months depending on whether the Member State Committee is involved or not. Thus under favourable conditions, first decisions are likely to be taken between the middle and end of the following year after CoRAP inclusion. The registrants will have the opportunity to comment on the draft decision before the final decision is taken.
The decision will define the deadline by which the registrant(s) must provide the necessary information. Depending on the type of information, the deadline may be between some months and several years.
"Follow up" under substance evaluation means: once the requested information is available and evaluated by the Member State, it will consider whether and how to use the information obtained for the purposes of Community level risk management measures. The follow up can either be no action or recommendation to take further actions, such as to propose EU wide risk management measures.
A follow up conclusion under substance evaluation is not directly initiating further risk management measures. Any proposed Community-wide actions will be subject to a separate decision making process and Member States need to file a notification for this purpose. For authorisation, restriction and/or harmonised classification under the REACH and the CLP Regulations, stakeholders are consulted at all relevant stages of the process and decisions are taken on the basis of the opinions adopted by the ECHA Committees.
Information on the substances is available on the ECHA website. This website contains non-confidential information on the properties and uses of the substances that have been retrieved from the registrations for each substance.
Decisions on requests of further information and substance evaluation reports prepared by the Member States will also be published on the ECHA website, when they are available.
Dissemination portal:
http://echa.europa.eu/information-on-chemicals/registered-substances
CoRAP-substances:
https://echa.europa.eu/information-on-chemicals/evaluation/community-rolling-action-plan/corap-table
Where exposure to workers in the EEA is established, the REACH requirements apply. Note that the REACH provisions under Annexes VII to XI encourage the use of adaptations; animal testing should be performed as a last resort only (Article 25 of REACH).
Consequently, testing on vertebrate animals will be required only if there is no available information which meets the information requirements, and where no adaptation possibility under column 2 of REACH Annexes VII to X, or under Annex XI can be applied.
This will represent the only means to assess the potential human health risks arising from exposure to workers.
As for every adaptation of an information requirement in a registration dossier, you need to insert a justification in each of the relevant endpoints of the IUCLID dossier.
Two main types of scenarios have been identified:
- Where the substance is imported into the EEA in a cosmetic product that is not further processed in the EEA: in addition to following the instructions provided by ECHA (see below), you shall add, to the respective endpoint(s) in IUCLID, an explanatory note stating that the substance is solely used in cosmetics, imported in the finished state and not further processed nor repackaged inside the EEA;
- Where the substance/cosmetic product is further processed in the EEA, but where absence of worker exposure can be demonstrated: you may avail yourself of the regular adaptation possibilities, pursuant to Annex XI, section 3.1 of REACH, to waive the testing requirements addressed by sections 8.6 and 8.7 (repeated dose toxicity and reproductive toxicity respectively) of Annex VIII to REACH and the test in Annex IX and X.
When applying these adaptations, for the purpose of the justification required according to Annex XI, section 3.2, you do not need to consider the life-cycle stages related to the use of the finished cosmetic product, as these are regulated separately under the Cosmetics Regulation.
While testing for acute toxicity cannot normally be waived under Annex XI, section 3.2, for the purpose of registrations dossiers that cover only cosmetic uses, a similar waiver containing the elements of Annex XI, section 3.2 may be used for this endpoint.
See section 5.1 of the Guidance on information requirements and chemical safety assessment, Chapter R.5: Adaptation of information requirements for further details on how to make use of this adaptation possibility.
Furthermore, ECHA provides further specific recommendations to follow below when you create or update your registration dossier.
In IUCLID 6, a request for ‘waiving' a standard information requirement under REACH must be recorded in the fields ‘Data waiving' and a ‘Justification for data waiving' must be recorded for each endpoint where waiving is proposed. Please follow the instructions below when you create or update your registration dossier.
The description of the information to be provided has been organised by ‘IUCLID Section' and ‘Field'. In addition, a distinction has been made between the information to be entered in the substance dataset, and information that can only be entered when you create the dossier.
Please use from the column "Selection/ entry" the appropriate pick-list selection and the recommended standard text to be entered in the corresponding ‘Field'.
If the substance is only imported in the EEA in a cosmetic product in its final state (neither further processed nor repackaged inside the EEA), the waiving possibility is only relevant for human health endpoints and is only based on the fact that there are no uses in any stage of the life-cycle which may be relevant to REACH (no exposure to workers; exposure to professionals and consumers is covered by the Cosmetics Regulation).
The following specific data waivers can be used only for human health information requirements (Sections 8 of REACH Annexes VII-X).
|
IUCLID section |
Field |
Selection / entry |
||
|
Substance dataset |
Any Endpoint Study Record– Annexes VII to X * |
Data waiving |
’study waived due to provisions of other regulation’ |
|
|
Justification for data waiving |
‘other:’ + “The substance is used exclusively in cosmetic products which fall within the scope of the Cosmetics Regulation. See field ‘Justification for type of information’ for further details.” |
|||
|
Justification for type of information |
“The substance is used exclusively in cosmetic products which fall within the scope of the Cosmetics Regulation. The substance is imported in a cosmetic product in its final state (i.e. the product is from the time of import neither further processed nor repackaged inside the EEA). Waiving of animal testing requirements for human health endpoints is proposed based on the absence of uses other than in finished cosmetic products.” |
|||
|
Section 3.5.5 – Consumer uses ** |
Product category *** |
PC39 |
||
|
Dossier |
Dossier header |
Dossier submission remark |
“This dossier covers a substance that is used exclusively in cosmetic products which fall within the scope of the Cosmetics Regulation. The substance is imported in a cosmetic product in its final state. The product is from the time of import neither further processed nor repackaged inside the EEA.” |
|
|
Updates only |
Dossier header |
Spontaneous update, ‘Justification' **** |
‘other' + ”Cosmetics Regulation / 2013” |
|
* Data waiving can only be applied to the endpoints required by the REACH Annexes for the tonnage band corresponding to the registration dossier.
** The information to be provided for each use, entered in IUCLID section 3.5.5: Consumer uses, is described in the manual How to prepare registration and PPORD dossiers (Chapter 9.6.4.3 and Annex 2). The manual is available on the ECHA website (under Support > Manuals), as well as inside the IUCLID 6 Help system.
***All uses outside the cosmetic use have to be documented in Section 3.5
**** A separate justification should be entered for each reason for the update.
In IUCLID 6, a request for ‘waiving' a standard information requirement under REACH must be recorded in the fields ‘Data waiving' and a ‘Justification for data waiving' must be recorded for each endpoint where waiving is proposed. Please follow the instructions below when you create or update your registration dossier.
The description of the information to be provided has been organised by ‘IUCLID Section' and ‘Field'. In addition, a distinction has been made between the information to be entered in the substance dataset, and information that can only be entered when you create the dossier. Please use from the column "Selection/ entry" the appropriate pick-list selection and the recommended standard text to be entered in the corresponding ‘Field'.
If the substance is further processed inside the EEA, i.e. it is imported or manufactured in the EEA, and still further formulated or re-packaged, before or after inclusion in the final cosmetic product, you need to demonstrate the absence of exposure to workers to benefit from the adaptation possibility.
This case also covers situations where you do not need to provide an exposure assessment: either no CSR is required due to the low tonnage of the substance manufactured or imported, or no exposure assessment is required because the substance does not require classification.
Consequently, you should document the absence of exposure as appropriate, using exposure scenarios and/or other approaches. You can apply for the following specific data waivers, specifically for the human health information requirements (Sections 8 of REACH Annexes VII-X).
|
IUCLID section |
Field |
Selection / entry |
||||||
|
Substance dataset |
Endpoint Study Record corresponding to: * -Annex VII -Annex VIII (except sections 8.6 and 8.7) |
Data waiving |
‘study waived due to provisions of other regulation’ |
|
||||
|
Justification for data waiving |
‘other:’ + “The substance is used exclusively in cosmetic products which fall within the scope of the Cosmetics Regulation. See field ‘Justification for type of information’ for further details.” |
|
||||||
|
Justification for type of information |
“The substance is used exclusively in cosmetic products which fall within the scope of the Cosmetics Regulation. The substance is manufactured and/ or further processed inside the EEA before/ after inclusion in the final cosmetic product. Waiving of animal testing requirements for human health endpoints is proposed, since the substance is handled only under strictly controlled conditions, with the exception of the life-cycle stage that covers the use as a cosmetic product (for which the safety assessment is done under the Cosmetics Regulation). Where it is demonstrated that the substance is handled according to strictly controlled conditions (see, as an example, REACH Annex XI, Section 3(2)(b)), during all life-cycle stages, with the exception of the use as a cosmetic product, the absence of exposure to workers is documented in IUCLID section 13:”
|
|
||||||
|
Endpoint Study Record corresponding to: * -Section 8.6 and 8.7 of Annex VIII |
Data waiving |
‘exposure considerations' |
|
|||||
|
Justification for data waiving |
‘other:’ + “The substance is used exclusively in cosmetic products which fall within the scope of the Cosmetics Regulation. See field ‘Justification for type of information’ for further details.” |
|
||||||
|
Justification for type of information |
“The substance is used exclusively in cosmetic products which fall within the scope of the Cosmetics Regulation. The substance is manufactured and/ or further processed inside the EEA before/ after inclusion in the final cosmetic product. Waiving of animal testing requirements for human health endpoints is proposed since the substance is handled only under strictly controlled conditions, with the exception of the life-cycle stage that covers the use as a cosmetic product (for which the safety assessment is done under the Cosmetics Regulation). Where it is demonstrated that the substance is handled according to strictly controlled conditions (see, as an example, REACH Annex XI, Section 3(2)(b)), during all life-cycle stages, with the exception of the use as a cosmetic product, the absence of exposure to workers is documented in IUCLID section 13:”
|
|
||||||
|
Section 3.5.5 – Consumer uses ** |
Product category *** |
PC39 |
|
|||||
|
|
||||||||
|
Dossier |
Dossier header |
Dossier submission remark |
“This dossier covers a substance that is used exclusively in cosmetic products which fall within the scope of the Cosmetics Regulation. The substance is manufactured and /or further processed inside the EEA before/ after inclusion in the final cosmetic product. All manipulation of the substance outside the cosmetics use takes place under strictly controlled conditions.” |
|
||||
|
Updates only |
Dossier header |
Spontaneous update, ‘Justification' **** |
‘other:' + “Cosmetics Regulation / 2013” |
|
||||
*Data waiving can only be applied to the endpoints required by the REACH Annexes for the tonnage band corresponding to the registration dossier.
**The information to be provided for each use, entered in IUCLID section 3.5.5: Consumer uses, is described in the manual How to prepare registration and PPORD dossiers (Chapter 9.6.4.3 and Annex 2). The manual is available on the ECHA website (under Support > Manuals), as well as inside the IUCLID 6 Help system.
***All uses outside the cosmetic use have to be documented in Section 3.5.
**** A separate justification should be entered for each reason for the update.
See also: https://echa.europa.eu/documents/10162/13628/reach_cosmetics_factsheet_en.pdf
Yes, the ECHA decision is legally valid and binding, so you have to comply with it.
However, if the substance is used exclusively in a cosmetic product and falls under one of the scenarios described, i.e. animal testing would only serve the purpose to address human health risks resulting from the exposure to the finished cosmetic product, you should be able to comply with the decision you received by requesting use of waiving possibilities, as per the REACH Annexes.
Illustration of cosmetics-based waiving scenarios.
It is only where the required testing relates to potential human health effects for workers that animal tests may be required. In such circumstances, the tests are performed to meet the requirements of REACH.
No, the entry into force of the total marketing ban under the Cosmetics Regulation (EC) No 1223/2009 does not influence the REACH requirements.
However, if your registered substance is exclusively used in cosmetics, ECHA recommends that you spontaneously update your registration dossier to clearly indicate the uses, should you wish ECHA to take this into account in any subsequent examination. Please follow the instructions provided according to the scenarios described in Q&A 991.
If you have not registered your substance already, ECHA recommends that you follow the instructions provided according to the scenarios described in Q&A 991.
- For more information regarding the Cosmetics Regulation and the requirements therein, please visit the European Commission website
- For more information on the interface of REACH and the Cosmetics Regulation, you may consult the factsheet ECHA has published in consultation with the European Commission.
- You may also contact the ECHA Helpdesk.
At an early stage, the co-registrants can already agree that one company takes over the organisation of the information exchange and the preparation of the joint submission. This is not, however, compulsory, as REACH does not set any conditions in this respect.
Where the information for exchange is considered commercially sensitive by one or more potential registrants (e.g. because of an impurity content that can indicate a production process), they can, for example, propose a confidentiality agreement or the use of an independent third party or trustee who can handle the confidential information on behalf of the potential registrants. Any other form of organisation is equally possible, as long as it is agreed by all existing and/or potential new co-registrants.
Detailed information on how to organise and improve communication between co-registrants can be found in the Guidance on data sharing.
There are several possible ways in which companies can organise their cooperation under REACH. These forms of cooperation can vary from loose ways of cooperating (e.g. IT tools to communicate between all members of a joint submission) to more structured and binding models (e.g. consortia created by means of contracts).
Participation in a SIEF (substance information exchange forum) was mandatory for phase-in substances until the last REACH registration deadline of 31 May 2018. After the end of the phase-in period, however, the Commission Implementing Regulation 2019/1692 of 9 October 2019 encourages co-registrants to use similar informal communication platforms to enable them to meet their continuing registration and data sharing obligations under REACH and Commission Implementing Regulation 2016/9.
Membership of a consortium is, on the other hand, entirely voluntary. If some or all participants of one or more informal communication platforms decide to form a consortium, they are free to decide the arrangements regarding scope, purpose, duration, conditions for membership or leaving etc., as long as these do not contravene EU competition rules.
Additional information on forms of cooperation can be found in the Guidance on data sharing.
When a registrant does not own a study report that they require for their registration, they need to agree with its owner on the conditions of using the study report for REACH registration purposes. The owner of the data and the registrant are free to define the rights that will be granted.
If the robust study summary of a study has already been submitted to ECHA, a registrant can, for instance, refer to that study in their dossier, provided that they have permission to do so. In that context, the registrant and the data owner must agree on the conditions of the right to refer. The LoA is a term often used to describe the agreement on the sharing of data and granting a right to refer. The intellectual property rights of the data owner must in any case be respected by the potential registrant.
It is the common responsibility of the potential and the existing registrant(s) to negotiate and agree on the content of the joint registration dossier, submitted by a lead registrant. In the case of information submitted less than 12 years prior to the inquiry, Article 27 of the REACH Regulation requires that both potential and previous registrants make every effort to reach an agreement on the sharing of the data and its costs in a fair, transparent and non-discriminatory way. You can find here practical advice on data sharing negotiations.
In case you fail to reach such an agreement, you may, as a last resort measure, contact ECHA in accordance with Article 27(5) of the REACH Regulation. Please find more details under point 5 here.
Potential registrants may become aware that the previous registrant who submitted the data they need for registration purposes has been included in a list of entities subject to restrictive measures or is otherwise subject to such measures. The sanctions in place may include a prohibition to make economic resources available to the sanctioned party, which may ultimately prevent the conclusion of a data sharing agreement. If the potential registrant believes to have made every effort to achieve an agreement, they may, as a last resort, submit a data sharing dispute with ECHA. To read more about data sharing disputes, check our Guidance on data sharing.
For more information on the impact of the sanctions, please see the FAQs published by the European Commission at this link.
- Show your real intentions to register or update your registration dossier, by providing ECHA with the registration numbers or inquiry numbers and the EC number of the substances you manufacture or import;
- Provide a description of why you need the data and how you intend to use it (e.g. for read-across);
- Submit a declaration that "The robust study summaries provided shall be only used for registration purposes under the REACH Regulation";
- Provide the EC numbers of the substances for which you need the robust study summaries submitted more than 12 years previously
- List the endpoints that you would require information for.
According to REACH, any robust study summaries submitted more than 12 years previously can only be used for the purposes of registration. Furthermore, the provision by ECHA of copies of robust study summaries does not provide ownership rights on this data.
- When potential registrants are informed of the previous and other potential registrants (and vice versa) by ECHA following an inquiry pursuant to Article 26 REACH;
- After the successful submission of the registration dossier, whenever new information becomes available. In such cases, according to Article 22 REACH, registrants have to update the joint registration dossier. This may require prior data sharing and may have an impact on decisions on the classification and labelling. It can also lead to the need to change the chemical safety report;
- As a consequence of dossier evaluation by ECHA (compliance check or the assessment of a testing proposal) or substance evaluation. These processes may lead to a request to submit further information, which would need to be addressed among all registrants of the same substance. For substance evaluation cases, these are not necessarily limited to the tonnage band related information requirements. Registrants should agree on the generation of requested information and on the sharing of the data and costs. In certain cases, also registrants who ceased manufacture may be requested to provide additional information. Therefore, data sharing does not only apply to "existing" studies but also to studies which will be needed to ensure that the registration is compliant with REACH.
The substance has been registered but the relevant studies were submitted less than 12 years before the inquiry. ECHA informs the potential registrant of the names and addresses of the previous registrants.
If he needs information involving tests on vertebrate animals, the potential registrant must contact the previous registrants (and/or the other inquirers) identified by ECHA. If he needs information not involving tests on vertebrate animals, the potential registrant may contact these previous registrants (and/or the other inquirers).
A request for sharing information must be made for any studies involving vertebrate animals. However, a potential registrant has to request data from the previous registrants if that data does not involve testing on vertebrate animals. The previous registrant and the potential registrant have to make every effort to reach an agreement on the sharing of the data requested and its costs. They must also apply the obligations set out in the Implementing Regulation 2016/9 on joint submission and data sharing. The obligations to make every effort and those set out in the Implementing Regulation apply to any information requested, whether it concerns vertebrate or non-vertebrate animal studies.
See Q&A 102 for access to data submitted more than 12 years previously.
The situation:
After an inquiry submitted under REACH Articles 26 (inquiry for a substance before its registration) or 12(2) (inquiry for a tonnage band increase), the previous registrant (be it the lead or a member) may, pursuant to Article 27(8), ask ECHA to extend the registration waiting period by an additional four months. Accordingly, the inquirer upon receipt of confirmation of the successful registration, will have to wait for an extra period of four months before being entitled to manufacture or import the substance (in a higher tonnage band, if relevant) on the European market.
The process:
The previous registrant can make the request to ECHA Helpdesk by using the web form and should include the following information:
- Statement "Request for Extension of the registration waiting period under REACH Article 27(8)"
- The inquiry submission number (such as AB123456-78)*
- The EC/List/CAS number of the inquired substance*
*both are included in ECHA’s REACH-IT messages received by the previous registrant following an inquiry.
As a result, ECHA will inform the potential registrant, through REACH-IT, that the previous registrant requested to extend the registration waiting period by an additional four months.
If the previous registrant intends to request an extension of the registration waiting period, they should contact ECHA as soon as they become aware of any new successful inquirers, given that registrations can be issued, in some instances, within a relatively short period of time.
The objectives of data-sharing are to avoid the duplication of work and in particular to reduce testing involving animals. These objectives are still fully relevant after the end of the phase-in scheme. Therefore, it is in line with the objectives of data sharing under REACH, as well as in the interest of potential registrants and in your own interest (as you will get compensated) to share data with potential registrants.
The use of published data to satisfy your information requirements requires you to have the right to refer to the full study report. Therefore, if you want to refer to a published full study report in your registration dossier, you have to check with the copyright owner to what extent you are allowed to use it in your own dossier.
In this case, you should negotiate a license or other form of agreement (e.g. letter of access) that will allow you to refer to the published data. For efficiency reasons, such an agreement should ensure that all the members of the joint submission have the right to refer to the data.
Copyright covers only the form of expression but not the facts and data included in the work. Therefore, facts and data can be included in the dossier without the consent of the copyright owner provided that the text of the study is not copied as such into your registration dossier. In other words, you can use the data to produce your own study summary but you have to make appropriate references and quotations to the original study to acknowledge the source of information. In addition, also in cases where you produce the study summary yourself, you must have the right to refer to the full study report for your registration. For more information, see the Guidance on data sharing, section 3.3.3.8.
All potential registrants of a substance must examine all available information and apply the relevant classification and labelling for their substance as it has been mutually agreed within their SIEF.
If they disagree with this classification, they may decide to ‘opt-out’ from this information requirement and propose a different classification, as long as they appropriately justify their decision (Article 11(3)(c), REACH).
Nevertheless, potential registrants of a substance in a low tonnage band do not have to contribute to the cost of a study if this study is not required for a registration in their tonnage band (Article 11(2), REACH).
There is no formal procedure to get in contact with the joint submission of another substance for read-across purposes. You can look up the details in our dissemination portal for registered substances, and contact any of the registrants whose name appears, asking for the lead registrant’s contact details. You can then contact them in order to find a data sharing agreement.
Alternatively, you can use our contact forms and request ECHA to share your contact details with the lead registrant of the joint submission of interest. We will then contact the said lead registrant and encourage them to contact you.
Please note that data sharing between different substances for read-across purposes is not mandatory. However, the Commission Implementing Regulation (EU) 2016/9 of 5 January 2016 on joint submission of data and data sharing underlines the benefits of data sharing between different substances. Therefore, data sharing between similar substances is strongly recommended, but it cannot be imposed by ECHA.
No, under REACH, a third party representative (TPR) cannot register. A TPR can be appointed by a manufacturer, importer or downstream user for data sharing issues and discussions with other manufacturers, importers or, where relevant, downstream users. Unlike an only representative, a TPR only plays a part in the negotiations between the (potential) registrants, while the appointing company remains responsible for complying with its registration obligations.
If a (potential) registrant changes its Third Party Representative (TPR), the new TPR will have to create a REACH-IT account (provided he does not have one yet).
In case of a change of TPR, the following steps have to be taken by the (potential) registrant in order to update the details of its TPR:
To update a TPR in a registration submission: Go to menu -> Search -> Reference numbers -> Input the number in the search criteria -> Click on the results -> Click on Edit under Third party representative -> Add the new TPR.
No fee is required if the TPR is updated in a registration dossier.
No. The identity of the registrant will be published unless a confidentiality claim on the identity of the registrant is included in the registration dossier and the justification accepted as valid by the Agency.
A third party representative (TPR) can only be used to represent potential registrants in discussions with other manufacturers, importers and downstream users on data sharing and the joint submission of data as specified in Article 4 of REACH. The identity of the TPR is not published on the ECHA website. It will only be visible in:
- the potential registrants tab in the REACH-IT co-registrants page, if included in a successful inquiry notification;
- the registrants tab in the REACH-IT co-registrants page, if included in a successful registration dossier;
- and in the joint submission, if included during creation (lead registrant) or confirmation (member registrant) of the joint submission in REACH-IT.
You can nominate a Third Party Representative (TPR) to represent you in the joint submission discussions and in data sharing activities. This will keep your company name confidential vis-à-vis other co-registrants.
NB: The manufacturer or importer legally remains the registrant. The TPR must not be confused with the third party holding information (i.e. data holders), nor with an Only Representative. The TPR must also not be confused with the possibility to keep confidential the registrant’s name for registration purposes (Article 10(a)(xi) REACH).
For more information, see the Guidance on data sharing, section 3.2.3.1. Q&A 352 explains how to assign a TPR in REACH-IT.
During the Inquiry process: We display on the co-registrants page in REACH-IT the contact details of the TPR to registrants and successful inquirers. However, we communicate the result of the inquiry process to the inquirer and not to a designated TPR.
After the Inquiry result/ after Registration: It is also possible to appoint a TPR acting on behalf of the registrant after having registered a substance for data-sharing negotiations. You can appoint or change your TPR by updating your registration dossier. See Q&A 0063.
A TPR ensures that the potential registrant does not appear on the co-registrants page. To ensure confidentiality of the same company after the registration number has been granted, see Q&A 401.
Yes, they can, and are encouraged to do so if they hold significant data that will be valuable for registrants when compiling their registration dossiers. In practice, downstream users have to submit certain information (substance identification, contact details) to ECHA to be recognised as a data holder for a substance, after which ECHA puts them in contact with the potential registrants.
Data holders are entitled to be compensated for their data used for registration. More detailed information is available in the Guidance on data sharing, Section 3.2.3.2.
Consider taking specific measures in the SIEF to protect information that you consider CBI, but you nevertheless need to share with the SIEF to conclude on the substance sameness. You can, for example:
- Have confidentiality agreements that limit access to documents or other information to specific named persons, or departments; and
- Allow access to certain documents in a ‘reading room’ only (where copying is not allowed); and
- Agree to have certain documents reviewed and/or assessed only by a third party expert (independent consultant) or a trustee.
You can strengthen this by having additional personal confidentiality agreements for those who get access to the CBI documents.
As a minimum, you should specify to the other SIEF members that the information is indeed CBI and, therefore, you communicate it and it can be used only for purposes of the verification of substance identity under REACH.
For more information on CBI, see the Guidance on Data-sharing, section 9.
Downstream users can use substances, irrespectively of whether they have been registered or not. In this regard use means any processing, formulation, consumption, storage, keeping, treatment, filling into containers, transfer from one container to another, mixing, production of an article or any other utilisation. Placing on the market is however not to be regarded as a use. See Q&A 40 for further information.
Please note that for the use of substances (whether registered or not) certain requirements related to restrictions, authorisation and risk management may apply. Guidance on how to comply with these requirements is provided in the Guidance for downstream users available on the ECHA website.
The carriage of hazardous substances and mixtures by rail, road, inland waterway, sea or air is exempted from the scope of the REACH Regulation (see Article 2(1)(d)). Transporting activities (including loading and unloading) by transport companies are not "uses" under REACH.
The loading and unloading operations performed by the workers of the transport company are covered by the Carriage of Dangerous Goods legislation, and hence they are outside of the scope of the REACH Regulation. Compared to that, the site-related activities before loading and after unloading will often be "uses" under REACH, which may need an exposure scenario and a chemicals safety assessment.
It is also important to note that the transfer of substances and mixtures occurring exclusively within an industrial plant is covered by REACH, even if this includes transportation carried out by an external company.
Downstream users may make uses known to the suppliers in their supply chain, before the manufacturer or importer submits his registration, with the aim of making these uses identified uses. This right is enshrined in Article 37(2) of the REACH Regulation. When registrants base their assessment on information from downstream users, this helps to ensure that the advice they receive is directly applicable and that the handling of exposure scenarios is easier.
The information to registrants flows most efficiently through sector organisations, many of whom are developing use maps that describe the typical uses of their sector. Use maps describe the typical uses and conditions of use in an agreed template. Downstream users should check whether their sector organisations are preparing a sector use map that covers their use(s). Individual companies can also use these templates if they need to communicate any niche applications to registrants.
If a substance is subject to authorisation (Annex XIV):
- You must use the substance according to the conditions laid down in the authorisation granted for that specific use to an actor up your supply chain or apply for an authorisation yourself if the authorisation of your supplier does not cover your use(s);
- You must notify to ECHA within 3 months after first supply, the use of the substance subject to authorisation.
If a substance is subject to restrictions (Annex XVII):
- Comply with the restrictions for placing on the market or use of substances as listed in Annex XVII of the REACH Regulation.
Suppliers must include information on authorisation and restriction in Section 15 of the safety data sheet or in other information provided in accordance with Article 32 of the REACH Regulation.
If your suppliers are located outside the EU and decide to appoint an only representative, they will confirm this to all the importers. You should preferably also obtain confirmation in writing from the only representative that your imported tonnage and use is indeed covered by the registration dossier.
This would not only provide you with a contact point with whom you can make your use known, but would also clearly document that your imports are indeed covered by the registration of the only representative.
You need to keep exact documents on which imported quantities of the substance are covered by the only representative registration and which imported quantities are not. For further information see the Guidance on registration, chapter 2.1.2.5 "Only representative of a non-EU manufacturer".
If a safety data sheet (SDS) is required for your substance, you will continue to receive it. However, when the SDS is updated after registration, you will see the registration number under section 1.1. You should also notice a change in that the updated SDS may contain one or more exposure scenarios as annexes, if your supplier has registered the substance for 10 tonnes/year or more. These exposure scenarios outline the conditions of safe use of the substance for specific uses.
You need to identify which exposure scenario(s) apply to your use(s) and check whether your conditions of use are in line with them. You will also need to take this information into account when communicating on safe use for the products that you place on the market.
Note that the exact time for updating the SDS is not defined under REACH and will depend, among other things, on whether any new information on the hazards and risk management measures have been generated in the course of the registration process.
According to the legal text the 12 months starts as soon as the DU receives an SDS containing a REACH registration number (Article 39(1) REACH). Nonetheless, it is understood that the DU requires an ES to be attached to the SDS, or at least for “uses advised against” to be included in Section 1 of the SDS, in order to determine if their uses are indeed included or excluded in the registration dossier. In cases where the required information has not been provided in the SDS, it is advisable that the DU communicates with his supplier to check why, record this communication, and the date when they receive an ES.
If a downstream user uses the substance (as such or in a mixture) under conditions that are not communicated by the supplier in the extended safety data sheet (eSDS), or the use is not covered at all in the eSDS, they may choose one of the following options:
- Ask your supplier to include your use or conditions of use in their chemical safety report and to provide you with an exposure scenario for it. You need to make sufficient information available to your supplier to enable them to carry out your Chemical Safety Assessment (CSA). Your sector organisation may have developed a convenient means of supplying this information specifically to your sector.
- Implement the conditions of use described in the exposure scenario you have received. This option may require an adaptation of your processes to meet the conditions described in the eSDS.
- Substitute your substance with a safer alternative.
- Find another supplier who can provide the substance with a safety data sheet and an exposure scenario covering your use.
- Carry out your own chemical safety assessment and prepare your own downstream user chemical safety report (DU CSR) for your uses and conditions of use, unless exemptions apply – see our practical guide How to prepare a downstream user chemical safety report for details. Notify your use, including the information specified in Article 38(2) of the REACH Regulation to ECHA.
If a downstream user holds information that puts into question the hazard or risk management information received from the supplier, they should communicate such information upstream to the supplier.
For further information, we advise to consult the Guidance for downstream users:
- An overview of how to decide whether or not your use is covered by the exposure scenario can be found in Section 4 Downstream users and exposure scenarios
- Information on how to make a downstream user chemical safety report is given in Section 5 Use not covered: preparing a downstream user chemical safety report (DU CSR)
More information is also available in the Downstream user responsibilities section and the practical guide How downstream users can handle exposure scenarios (section 2.2.2 What to do if the use and/or conditions of use are not covered by the exposure scenario).
You have to report to ECHA within 6 months of receipt of the safety data sheet for a registered substance when you:
- Need to prepare a downstream user chemical safety report; or
- Wish to benefit from the exemption to prepare a chemical safety report either because:
- you use the substance in total less than 1 tonne per year; or
- You use the substance for product and process oriented research.
You also have to report to ECHA if your classification of a substance differs from that of all of your suppliers. Reporting is not required if the downstream user uses the substance or mixture in a total quantity of less than one tonne per year.
If you use a substance included in the Authorisation List, for which an authorisation has been granted that covers your use, you have to notify ECHA of your use within three months of the first supply of the substance.
A downstream user chemical safety report documents the results of the chemical safety assessment undertaken by the downstream user. The assessment establishes conditions to ensure that the risk for the use(s) not covered in the received exposure scenarios is adequately controlled. The downstream user chemical safety report itself does not need to be submitted to ECHA.
A downstream user report is a report by a DU to ECHA when:
- He has to prepare a downstream user chemical safety report or is claiming exemption
- His classification of the substance is different to that of his supplier
- A downstream user notification is required when a downstream user uses a substance included in the Authorisation List, for which an authorisation has been granted that covers the use.
A downstream user can make a testing proposal. ECHA examines this proposal and decides on appropriate testing in accordance with Article 40 of REACH. It is the responsibility of the downstream user to perform the test, unless other downstream users or registrants are also interested in carrying out such a test.The interested parties can agree on who will perform the test on behalf of all of them. If agreement is not reached, the Agency shall designate one of the parties to perform the test on behalf of all. All parties concerned share the cost of the study.
- Option (i): The webform is the simpler option. It is recommended for most downstream users, especially those who have not used IUCLID before.
- Option (ii): Reporting via REACH-IT is recommended for downstream users who already use IUCLID and want to maintain all their report records in the REACH-IT/IUCLID system.Downstream users who need to report if their classification is different to that of their supplier can only use option (ii), via REACH-IT.
- the identity and contact details of the downstream user;
- the registration number of the substance, if available;
- the identity of the substance;
- the identity of the supplier;
- a brief general description of the unsupported use(s) and conditions of use; and
- a proposal for additional testing on vertebrate animals if this is considered necessary.
A tutorial is available on using the webform.
http://echa.europa.eu/view-article/-/journal_content/title/submitting-using-the-webform
- Prepare a Downstream user report dossier in IUCLID 6 as explained in the manual How a downstream user report
- In the IUCLID 6 dataset, enter information on your classification and labelling of the substance as explained in the manual How to prepare a classification and labelling notification
- Create the downstream user report dossier and export it as explained in the downstream user manual
- Submit the report to ECHA via REACH-IT
No fee is charged for the submission of a downstream user report. ECHA fees and charges for services and their amounts are stated in the REACH Regulation and in Regulation (EC) No. 340/2008 (Fee Regulation).
It is possible that a reference substance does not exist in the downloadable reference substance list. In this case, you will need to create the reference substance yourself. The reference substance needs to be included in the substance dataset. You can find more information on how to create a reference substance in section 6.1 of the manual How to prepare registration and PPORD dossier.
Downstream users have to provide all the information as required by Article 38 of the REACH Regulation.Section 1.3 of IUCLID is where you provide the supplier's registration number for the substance, as required by Article 38(2)(b). If the registration number is not available to you, you must provide a justification for this in the same section.
Additionally, when updating a downstream user report, you should also include in this section the downstream user report's reference number.
Therefore, it is advised to follow the instructions included in the manual How to prepare a downstream user report.
If as a downstream user you use the substance (as such or in a mixture) outside the conditions communicated to you in the extended safety data sheet (eSDS), or the use is not covered at all in the eSDS, you may choose one of the following options:
- Adapt your conditions of use to those described in the eSDS.
- Implement or recommend an exposure scenario which includes as a minimum the conditions described in the exposure scenario communicated to you. Make the use known to the supplier with the aim of making it an identified use based on the manufacturer's chemical safety assessment.
- Perform your own chemical safety assessment for that particular use and record it in a Chemical Safety Report - CSR (if the total amount used is 1 tonne/year or more). Notify your use, including the information specified in Article 38(2) of the REACH Regulation to ECHA.
- Switch to another supplier of the substance if that supplier covers your specific use in his eSDS.
If as a downstream user you receive information from your customers intended to make a use known you should forward this information to the supplier up the supply chain or assess if the use is covered in the existing exposure scenario for the mixture and eventually carry out your own downstream user Chemical Safety Assessment (CSA).
If as downstream user you hold information that puts into question the hazard or risk management information received from the supplier you need to communicate this information to the supplier.
An overview of how to decide whether or not your use is covered by the exposure scenario can be found in section 6-'Deciding if the use is not covered by the exposure scenario' in the Guidance for downstream users. Information on how to make a downstream user chemical safety report is given in Section 7-'Making a downstream user chemical safety report' of the same guidance available at the ECHA website: https://echa.europa.eu/guidance-documents/guidance-on-reach
The obligations under REACH for a DU using a substance for the purpose of PPORD may differ, depending on whether or not the PPORD activity is covered by a PPORD notification made by the manufacturer or importer of the substance.
A DU, who is listed in a PPORD notification submitted by the manufacturer or importer as one of the customers, operates under the responsibility of his supplier and must respect any conditions set in accordance with Article 9(4) of REACH and/or communicated to him by his supplier. If the DU stops using the substance for the purpose of PPORD and, by this, ends the cooperation with his supplier, he needs to inform his supplier, as the supplier will need to update his notification to remove the DU from the list of customers.
Alternatively, a DU can use a substance for the purpose of PPORD under his own responsibility and initiative. Since a DU does not have the registration obligation of Articles 5 and 6 of REACH, there is no need for the DU to submit a notification under Article 9 of REACH to be exempted from the registration obligation.
If he adequately controls the risks to human health and the environment in accordance with the requirements of legislation for the protection of workers and the environment, the DU is not required to prepare a DU CSR, even if his conditions of use are not covered in the extended SDS of his supplier or the use is advised against (Article 37 (4) (f)). According to Art 38(1)(b), the DU must report to ECHA if using a registered substance at greater than 1 tonne for the purposes of PPORD and availing of the exemption in Art. 37(4)(f).
Manufacturers and importers of a substance on its own or in a mixture are encouraged to communicate with the downstream users or distributors of the substance with regard to whether and by when they intend to register the substance to enable the downstream user or distributor to seek alternative sources of supply if necessary. Once the substance has been registered, there is an obligation for the supplier to communicate the registration number down the supply chain either in the safety data sheet according to Article 31 or, if applicable, according to Article 32 of REACH.
A downstream user can check the registration status (see Q&A 399) of the substances on their own or in a mixture they place on the market, in order to comply with the obligation imposed by Article 5 of REACH to place on the market only substances that comply with the registration requirements under REACH.
If a downstream user suspects that their supplier should have already registered the substance, they should contact the supplier immediately. They should make sure that substances critical to their business are registered and that their uses are covered. Note also, that there may be a valid reason as to why your supplier has not registered the substance, e.g. the tonnage is below 1tpa or the substance is exempted (Annex V) from REACH. For further information, see our list of issues affecting downstream users.
The Integrated Regulatory Strategy, developed in consultation with the Member State competent authorities, aims to encourage registrants to comply with REACH and to improve their chemical safety assessments.
The Integrated Regulatory Strategy has three main pillars: 1) efficient selection of substances which may raise potential concern; 2) generation of the necessary information through compliance check for assessing their safety; and 3) assurance that any remaining concerns are subsequently addressed through the most suitable regulatory risk management instrument.
It can therefore be assumed that the selected substances that raise potential concern will be more likely addressed with further regulatory risk management measures, and that the substances that demand most urgent action will be part of the prioritisation.
More information is available at the Integrated Regulatory Strategy -page.
- high tonnages;
- suspected data gaps in the higher-tier human health or environment information requirements;
- high potential for exposure of humans or environment (e.g. widespread uses or uncertain information on uses).
If your dossier meets the criteria for selection (see Q&A 647), ECHA will start a compliance check assessment, focusing on selected information requirements (or properties) (see Q&A 654).
When a compliance check for a substance is launched, ECHA informs concerned registrants via REACH-IT. This notification also reminds the registrants of their legal obligation to keep the registration dossier up-to-date (see Q&A 1572).
At the same time, the information on the dossier evaluation status page will be updated. From then on, you can follow the progress of the evaluation through the entire process (see Q&A 1572) or via the ‘ECHA Dossier Evaluation status’ section of the relevant joint submission page in REACH-IT.
If the information requirements (or properties) assessed are found to be non-compliant, the relevant members of the joint submission will receive a draft decision from ECHA.
No, by default ECHA assesses specific information requirements (or properties) (see Q&A 0654) primarily focusing on dossiers of substances manufactured or imported at more than 100 tonnes per year.
ECHA may also target its checks to different information requirements; then the decision will indicate the scope of the assessment undertaken.
REACH does not limit the number of compliance checks per dossier. Therefore you may receive multiple draft decisions on the same dossier.
The updated dossier containing the information requested needs to be submitted at the latest by the deadline set in the (adopted) decision (Article 22(2) of REACH). The updated registration dossier will be assessed after the deadline set in the decision has passed (Article 42 of REACH), on whether it contains the information requested in the decision.
This is however without prejudice to your legal obligation to update your dossier with any (new) relevant information “without undue delay” (Article 22(1)).
Under ECHA’s Integrated Regulatory Strategy (see Q&A 0646), the compliance check of dossiers submitted for substances manufactured or imported at more than 100 tonnes per year mainly focuses on eight key information requirements (or properties): genotoxicity, repeated dose toxicity, pre-natal developmental toxicity, reproductive toxicity, carcinogenicity, long-term aquatic toxicity, biodegradation and bioaccumulation.
The goal is to focus on the information requirements (or properties) that matter most for human health and the environment, with special emphasis on those which are related to the persistent, bioaccumulative and toxic (PBT) or carcinogenic, mutagenic or toxic to reproduction (CMR) properties of a substance.
More information on the strategy on compliance checks is available at compliance checks page.
ECHA informs active registrants via REACH-IT, when a compliance check for a substances in their portfolio is launched. This notification is a reminder of the legal obligation to keep the registration dossiers up-to-date, which is particularly important when a compliance check is initiated.
You can follow the progress of the evaluation via the ‘ECHA Dossier Evaluation status’ section of the relevant joint submission page in REACH-IT or through the Dossier Evaluation status webpage as explained below.
Please note that even if your substance appears under assessment, you may not necessarily receive a draft decision, since it is sent only to registrants that are concerned by the data gaps identified or since it is sent to a registrant for reasons specific to that registrant (a draft decision may be registrant specific in cases where the evaluation concerns substance identity information or endpoints from which a registrant has opted out).
Even if you do not receive a draft decision, you can continue to follow the progress of the substance through the Dossier Evaluation status webpage.
If you have received a draft decision, it means that there are non-compliant information requirements (or properties) in your dossier or in the jointly submitted registration dossier.
Check in the decision which members of your joint submission have received the draft decision, and get promptly in touch with them.
You have 30 days to provide your comments on the content of the decision. ECHA strongly recommends that you provide your comments collectively, in one consolidated set. ECHA only accepts comments submitted using the webform. A link to the webform is provided in the notification letter accompanying the draft decision.
For more information, see also the ‘Decision under dossier evaluation’ under recommendations to registrants and the practical guide ‘How to act in dossier evaluation’
- If the requested information already exists, you will need to agree on the costs and sharing of the existing data.
- If the requested information needs to be generated, you will need to:
- agree on the test to be performed, including on the testing material to be used as representative for all registrants;
- inform ECHA of who will perform the test; and
- share the costs and new data
For increased transparency on evaluation and other processes, ECHA has published supporting information on the following pages:
For more information, see also ‘Decision under dossier evaluation’ under the Recommendations to registrants and check the practical guide ‘How to act in dossier evaluation’.
As of January 2019, ECHA addresses its (draft) decisions to all registrants relying on non-compliant information requirements. Similarly, ECHA addresses its (draft) decisions on testing proposals to all those registrants intending to rely on the proposed tests to fulfil their information requirement.
Substance information exchange forums (SIEFs) ceased to exist as of 1 June 2018. Nevertheless, the registrants of the same substance continue to be bound by the obligation to submit jointly the information on their substance.
By setting out clearly how the information requirements apply at each tonnage band, ECHA’s decision gives the registrants greater certainty and clarity on their individual legal obligations, which helps ensure that all registrants within the joint submission become compliant.
Addressing evaluation decisions to all non-compliant registrants also aims to supports data and cost sharing as well as collaboration and communication between co-registrants regarding their joint submissions.
All recipients must comply with their information requirements, and will need to share existing/new data while respecting their legal obligation to avoid unnecessary (vertebrate animal) testing.
ECHA’s dossier evaluation draft decision is based on the version of the registration dossier available in ECHA’s database at the time the draft decision is issued to the registrants for comments. ECHA does not examine updates of your registration dossier that have been submitted after this process step. Therefore, make sure that information on your tonnage, uses and registration type are regularly updated to match the actual use of the substance, so that ECHA can assess its safe use in Europe.
Your legal obligation is to keep your dossier up to date – in other words, to update your dossier without undue delay if you become aware of new information. Therefore, the initiation of a compliance check should not be a trigger for updating your dossier.
When ECHA starts the evaluation of a dossier, information on the dossier evaluation status can be found at Dossier evaluation status page. Furthermore, when a compliance check is launched, ECHA informs concerned registrants via REACH-IT (see also Q&A 1572). Therefore, you have the opportunity to verify and (if necessary) update your dossier especially regarding use and tonnage information before a draft decision is issued.
In the specific case where you cease manufacture or import of your substance after receiving the draft decision, please see Q&A 1580.
In the specific case where you update your dossier with substantial new information, including tonnage band updates, after receiving the draft decision, please see Q&As 1927 and 1928.
For more information, see also ‘Dossier evaluation decisions’ under the Recommendations to registrants and check the practical guide ‘How to act in dossier evaluation’.
The REACH Regulation sets the deadlines which apply during the decision-making procedure. The deadlines will not be extended, unless there are technical reasons (e.g. malfunction of the submission tools) or the commenting period falls during closure periods of the Agency.
Similarly strict timelines are in place for the proposals for amendment by Member State competent authorities, the referral to the Member State Committee (MSC), your comments on the proposals for amendment, and the agreement-seeking on the draft decision by the MSC.
For more details, see the practical guide ‘How to act in dossier evaluation’.
If you decide to cease manufacturing or importing your registered substance upon receipt of the draft decision, you should inform ECHA of your intention using REACH-IT (see Q&A 1439). When you confirm that you have ceased/are ceasing manufacture or import, ECHA proceeds by invalidating your registration number in line with Article 50(3), after which you are not allowed to manufacture or import the substance into the EU/EEA market.
Consequently, you will not receive the adopted decision (if you have already received an adopted decision, see Q&A 1580).
If you start to manufacture or import the substance again, you will have to register the substance once more and you may have to share the costs accrued for the maintenance and update of the registration dossier due to an evaluation process or for other reasons.
For more information, see also the practical guide ‘How to act in dossier evaluation’ and ECHA’s Recommendations to registrants.
If after receipt of a draft decision you have updated your dossier with a lower tonnage band and you wish ECHA to consider the downgrade in the ongoing decision-making process, you need to inform ECHA in your comments on the draft decision, or if the commenting period has passed, in a communication to ECHA via the contact form. In that communication you should refer to the REACH-IT message which delivered the draft decision (in format CCH-D-XXXXXXXXXX-XX-XX/D). You also need to provide the annual volume of the substance that has been imported and/or manufactured over the preceding calendar year together with documentary evidence supporting this information. ECHA will take the elements available into account to determine whether the change of tonnage band has been substantiated and whether there is a need to amend the draft decision accordingly.
For information on downgrades after receipt of an adopted decision, please see Q&A 1580.
If the commenting period is over, but you have not yet received the adopted decision, you can inform ECHA of substantial new information that may have an impact on the decision by contacting ECHA via the contact form. In that communication you should refer to the REACH-IT message which delivered the draft decision (in format CCH-D-XXXXXXXXXX-XX-XX/D). In your communication you also need to reproduce the information contained in your update, explain why it was not submitted in your comments to the draft decision and explain how it affects the information requests in the draft decision. Substantial new information means:
- Recent experimental studies that became available to you after receipt of the draft decision and which address data-gaps identified in the draft decision.
- Tonnage band changes (for tonnage band downgrades the information referred to in Q&A 1927 needs to be provided).
- Registration type changes (you will need to comply with the conditions set out in Articles 17 and 18 of REACH).
No, ECHA cannot alter the deadline of the decision, because it is unanimously agreed by the representatives of the Member States.
ECHA acknowledges that technical difficulties in testing a substance may lead to delays or to the inability to provide the requested information by the deadline set in the decision.
In any case, we advise you to submit an update of your dossier by the deadline set in the decision, including any relevant explanations and evidence concerning the possible delay and the expected submission date. You should then update your dossier again as soon as the missing information is available.
However, if you believe that the technical difficulties prevent you from testing altogether, you can, on your own responsibility, adapt the standard information requirements (see Q&A 1064).
After the deadline, ECHA will check whether a dossier update is submitted and whether the information provided fulfils the information requirements. If during the follow-up evaluation ECHA finds that some or all of the requested information is missing, the national enforcement authorities will be informed.
No. ECHA considers that after 10 years of REACH implementation, there is broad knowledge in all industry sectors, and that they are well placed to support you.
However, if you have questions regarding a (draft) decision, you may submit your enquiry to ECHA using a contact form and we will assess your specific question.
No, ECHA cannot discuss the content of the adopted decision after it has been unanimously agreed by the Member States representatives.
If you have general questions on ECHA’s decisions or processes, you may submit a request to the ECHA Helpdesk and we will assess your concerns.
Yes. If you believe that you have scientific grounds to fulfil the requested information by adapting the requests in the decision, you may do so on your own responsibility.
Any adaptation must fulfil the specific rules outlined in Annexes VII to X or the general rules of adaptations specified in Annex XI to the REACH Regulation.
See also ECHA’s Recommendations to registrants.
Once the deadline set in the decision has passed and after ECHA has assessed whether the information you submitted is sufficient, ECHA will address the non-compliance through a new decision-making process according to Articles 50 and 51 of REACH.
This means that you will receive a new draft decision informing you that the information you submitted is not sufficient and that you still need to address the request in the original decision in order to fulfil the information requirements for the endpoint(s).
This decision has no deadline. Once the decision is adopted by the Member States, ECHA will inform the national enforcement authorities of the countries where the registrants are located.
If upon receipt of the adopted decision, you cease the manufacture or import of your substance, or if you downgrade the tonnage band of your registration, you will have to comply with the decision, regardless. See also Q&A 1586.
For more information, see also the practical guide ‘How to act in dossier evaluation’ and ECHA’s Recommendations to registrants.
See also Q&A 1574.
Check the end of the decision for the list of addressees of the decision and contact them. You will need to collectively agree on who will perform the test(s) (see Q&A 1583) and inform ECHA about this within 90 days of the adoption of the decision.
If no agreement is reached, ECHA will designate one of the registrants to perform the studies on behalf of all other addressees of the decision. For more information, see the practical guide ‘How to act in dossier evaluation’.
After you have collectively agreed on who will perform the test(s), you have to inform ECHA accordingly within 90 days from the receipt of the adopted decision (Article 53(1) of REACH).
Please use the link to the webform provided in the notification letter accompanying the adopted decision:
You must indicate a name for each request listed in the decision, even if it is the same for all requests. If you fail to do so, ECHA will have to designate one of the recipients of the decision, for each test lacking an identified registrant.
All recipients of the same decision are collectively responsible for fulfilling the requests in the decision that are relevant to their registered tonnage band.
After you receive the adopted decision, you need to agree collectively on who of the recipients is going to perform the requested studies. This is independent from the fact that the lead registrant is the one who submits the information “acting with the agreement of the other assenting registrants” (Article 11(1) of REACH).
The recipients of the decision are expected to agree on the testing to be performed as per their data (and cost) sharing obligations. You also need to agree on the material to be tested for each study requested, ensuring that it produces appropriate information and is representative of the registered substance as manufactured by all members of the joint submission.
You are also collectively responsible for the submission of the requested information by the (lead) registrant appointed to do so, on behalf of the other registrants of the joint submission.
If you do not update your registration by the deadline set in the decision, because you ceased manufacture, or import, or downgraded tonnage band after you received the adopted decision, ECHA will inform the Member State competent authorities and the national enforcement authorities of the country you are located in (see Q&A 1065).
Your authorities are responsible for any enforcement actions related to evaluation decisions according to your national legislation.
Any registrant receiving the adopted decision, because they are not compliant with their information obligations, remains legally responsible for providing the information requested in the decision (see Q&A 1583).
Consequently, a change of lead after you receive the adopted decision does not change the legal responsibility of the other recipients.
In addition, if ECHA identifies that the registrants have failed to submit and comply with the information requirements, it will notify the enforcement authorities of all the countries where the incompliant registrants are located. Any enforcement actions related to evaluation decisions will be decided according to their national legislation, where appropriate.
No. ECHA will only check whether a dossier update is submitted once the deadline set in the decision has passed. ECHA will then evaluate whether the submitted information (test or adaptation) fulfils the requests in the decision (see Q&A 1064). This is part of the follow-up evaluation process.
In other words, ECHA does not perform any evaluations before the deadline in the decision has expired.
Once the deadline set in the decision has passed ECHA will assess the dossier. In case the information requested in the decision are not submitted, ECHA will inform the Member State competent authorities and the national enforcement authorities of the respective Member State about the failure to respond to the request(s) in the decision.
When ECHA sends information to national enforcement authorities, the responsibility for handling the non-compliance with REACH is transferred to the national authorities, which then consider and decide on enforcement actions, where appropriate.
Consequently, ECHA will not evaluate any subsequent dossier updates unless the respective national authority asks ECHA to do so.
You should direct all questions to your national authorities.
ECHA appreciates that the current global emergency has affected several registrants in the EU. For this reason, ECHA extended certain deadlines in the registration and evaluation processes, for a limited period of time, to take account of the current global situation (see more here).
However, the Agency is not in a position to alter the final deadlines set in adopted decisions. This is because these are decisions that have been agreed in close consultation with the Member States.
As usual, ECHA will initiate the follow-up evaluation of the updated registration dossier for the substance subject to dossier evaluation process that you mentioned in your document when the deadline in the decision has passed. Then, ECHA will establish whether the new information submitted in an updated dossier corresponds to the requests in the decision.
If you think you will not be able to provide all the information requested in the decision by the deadline set, we advise you to anyway update your registration dossier by the deadline and, if necessary, include all relevant explanations and proof concerning the status of ongoing tests and the reasons for the delay, including the expected submission date. As soon as the missing information becomes available to you, you should update your registration dossier again.
According to Article 42(1), ECHA needs to examine all of the information submitted as a response to a testing proposal and compliance check decision. The Agency also has to draft any appropriate decisions, if necessary.
If the information provided in the dossier update does not fulfil the information requirements, the Member State competent authorities will be informed. They will then have to consider your argumentation and decide on any enforcement actions, where appropriate.
For further information on the follow-up to dossier evaluation decisions, please see the steps in the Evaluation process and the answers to the most frequently asked questions on Evaluation on the ECHA website.
When you perform new tests you have to follow the Test Method Regulation (Commission Regulation No 440/2008) or another method recognised by the European Commission or ECHA (Article 13(3) of REACH).
In REACH Annexes VII to X on standard information requirements, the use of various OECD test guidelines is required (e.g. OECD TG 414, 421 and 422).
The OECD methods can be found at: http://www.oecd-ilibrary.org/
The text of the Test Method Regulation can be found at: http://eur-lex.europa.eu/JOHtml.do?uri=OJ:L:2008:142:SOM:EN:HTML
Note that changes occurred in the Annexes VII and VIII requirements in 2016 for the following endpoints: skin and eye irritation, skin sensitisation and acute dermal toxicity, making non-animal testing the default.
Information may be generated using other methods (Article 13(3)) provided the conditions defined in Annex XI are met. These include amongst others that the result is sufficient for the purposes of classification and labelling and/or risk assessment, and that adequate and reliable documentation of the applied method is provided (see Annex XI for more information).
Furthermore, a specific requirement exists for ecotoxicological and toxicological tests: new tests have to be carried out in compliance with the principles of good laboratory practice (GLP) provided for in Directive 2004/10/EC, as no other international standard has so far been recognised as being equivalent. For physicochemical testing, it may be desirable but it is not mandatory to have tests performed according to the GLP standard.
The Guidance on Information Requirements and Chemical Safety Assessment contains specific integrated testing strategies for each endpoint (e.g. for aquatic toxicity, mutagenicity), which should be consulted before new tests are performed. You can find this document at: http://echa.europa.eu/guidance-documents/guidance-on-information-requirements-and-chemical-safety-assessment
Die Europäische Kommission hat die von den nationalen GLP-Überwachungsbehörden vorgelegten Listen von inspizierten Prüfeinrichtungen veröffentlicht. Sie können dieses Dokument abrufen unter
http://ec.europa.eu/DocsRoom/documents/8575/attachments/1/translations/en/renditions/native
Für die Zertifizierung von Labors nach den Grundsätzen der Guten Laborpraxis (GLP) sind die nationalen Behörden zuständig, die auch die nationalen Überwachungsprogramme verwalten.
Falls das Labor seinen Sitz in der EU, in Norwegen oder der Schweiz hat, ist die entsprechende Behörde auf der Website der GD Unternehmen und Industrie der Europäischen Kommission zu finden unter:
http://ec.europa.eu/growth/sectors/chemicals/good-laboratory-practice/index_de.htm
Befindet sich das Labor in einem anderen Land, konsultieren Sie bitte den Abschnitt zu GLP auf der Website der OECD:
http://www.oecd.org/chemicalsafety/testingofchemicals/goodlaboratorypracticeglp.htm
Nachdem Sie die zuständige GLP-Überwachungsbehörde ausfindig gemacht haben, können Sie sich bei dieser Behörde nach den Labors erkundigen, die in dem entsprechenden Land nach GLP zertifiziert sind.
Außerdem können Labors von einer GLP-Überwachungsbehörde auch dann inspiziert werden, wenn sie sich in einem Land befinden, das nicht dem OECD-System über die gegenseitige Anerkennung von Daten beigetreten ist. Auskünfte über diese Labors erteilt die GLP-Überwachungsbehörde, die diese inspiziert hat (siehe hierzu auch Q&A 122 ).
In general, there is the possibility to use data from reliable, scientifically accepted reference literature or databases, provided that the substance to be registered and the substance described in the reference are comparable with regard to homogeneity, impurities, particle size etc.
The documentation of similarity needs to be submitted in the registration dossier. References to literature or databases often use secondary data sources. When such data is used, the original source should be cited and checked by an expert.
Some useful reference books and data compilations containing peer reviewed data are listed under each endpoint in the Guidance on Information Requirements and Chemical Safety Assessment, Chapters R.7 a, b, c: Endpoint specific guidance available on the ECHA website at: http://echa.europa.eu/guidance-documents/guidance-on-information-requirements-and-chemical-safety-assessment.
For some endpoints, these data from reference literature or databases may be used on their own to fulfil the information requirement. However, in general they will have to be combined to other pieces of evidence and submitted as part of a weight-of-evidence approach or read-across approach to support the justification proposed to adapt the requirement.
The OECD decision on mutual acceptance of data (MAD) provides for data generated by testing of chemicals in an OECD member country in accordance with OECD test guidelines and OECD principles of good laboratory practice (GLP) to be accepted in other member countries for purposes related to the protection of human health and the environment.
This system also covers non-OECD countries, which have requested adherence to the OECD GLP and to join the MAD system. These non-OECD countries can be divided into two groups:
- Countries that are full adherents to the OECD MAD system.
- Countries that are provisional adherents to the OECD MAD system.
Countries that are full adherents to the OECD MAD system will accept data from OECD member countries and other adhering countries generated under MAD conditions. In addition, non-clinical safety data developed in these countries must be accepted by the OECD and adhering countries.
Countries that are provisional adherents to the OECD MAD system need to accept data from OECD member countries and other adhering countries generated under MAD conditions. However, during the period of provisional adherence, GLP monitoring activities conducted by the GLP monitoring authority located in the country of the provisional adherence do not have to be accepted by the full members of the OECD MAD decision.
http://www.oecd.org/env/ehs/mutualacceptanceofdatamad.htm
In general, ECHA accepts data as GLP data where this data comes from a test facility:
- from countries that are OECD member states or full adherents to the OECD mutual acceptance of data (MAD) system; and
- from countries that are provisional adherents to the OECD MAD system and in which laboratories have been inspected jointly by the GLP monitoring authority concerned and by an OECD GLP monitoring authority.
Studies that are conducted in a test facility situated in a country which has not joined the OECD MAD system can be accepted by ECHA as GLP compliant studies under the following conditions:
- Before performing the study, the GLP compliance of the test facility has been inspected by:
- an EU GLP monitoring authority (including Norway through EEA agreement); or GLP monitoring authorities in Israel, Japan and Switzerland with whom the EU holds mutual recognition agreements; or
- other GLP monitoring authorities of OECD member states or full adherents to the OECD MAD system on a case-by-case basis; or
- other national GLP monitoring authority which has been assessed on-site by representatives of the EU GLP Working Group and whose Compliance Monitoring Programme could be regarded as being equivalent to the EU GLP Compliance Monitoring Programme; and
- The test facility has been found to be operating in compliance with GLP principles.
http://www.oecd.org/env/ehs/mutualacceptanceofdatamad.htm
See also Q&A 119.
Not necessarily. The scope of the risk characterisation, that you have to carry out as part of the CSA, depends on the hazard profile of the substance. It has to address every hazard, not just those that lead to a classification (points 0.5 and 6 in Annex I to REACH).
Firstly, you have to consider each physical, health and environmental hazard identified, even if classification is not required. This includes collecting the predicted or derived no-effect levels or minimal effect levels (PNECs, DNELs or DMELs) if appropriate.
You should also consider the relevant timescales, environmental compartments, human populations, health effects, and routes of exposure.
DNELs for irritation/corrosion can only be derived if dose-response information is available. Therefore, for endpoints such as eye irritation where no DNEL can be derived, a more qualitative approach to assessing and controlling such risks is necessary. This may be the case where the pH led to the classification or where only QSAR data are available.
For information about this approach, see Chapters R.8 (Part E) and R.10 of the Guidance on Information Requirements and Chemical Safety Assessment: http://echa.europa.eu/guidance-documents/guidance-on-information-requirements-and-chemical-safety-assessment.
If there are no other hazards, then it is sufficient to describe the measures which ensure that the risks to eyes are avoided or managed in the exposure scenarios (ESs). If there are other hazards identified, then your assessment should address these also.
The exposure assessment and the subsequent risk characterisation should cover all stages of the life cycle of the substance resulting from the substance's manufacture, and the identified uses.
The Practical Guide on How to undertake a qualitative human health assessment and document it in a chemical safety report is a helpful document when undertaking a qualitative human health assessment: http://echa.europa.eu/practical-guides
Tip: Using Chesar will help you determine the scope of exposure assessment and the type of risk characterisation. For more details on this see the ‘Chesar User Manual, Part 1, Section 6' available at: http://chesar.echa.europa.eu/web/chesar/support/manuals-tutorials
For aquatic toxicity testing (Section 9.1 Annexes VII and VIII), Column 2 adaptations include two complementary concepts related to solubility in water.
The concept of "highly insoluble in water" is associated with the likelihood for aquatic toxicity; consequently a general threshold cannot be established. The use of this concept for waiving aquatic toxicity testing requires substance-specific assessment.
In the waiving statement, registrants should justify that aquatic toxicity is unlikely to occur at the limit of the water solubility. This may require specific information, such as that obtained from transformation/dissolution studies or from the identifying the components of the water accommodated fraction (see the webinar presentation: Hints and Tips on Physicochemical, environmental and human health related endpoints - Aquatic Toxicity).
If registrants cannot demonstrate that aquatic toxicity is unlikely to occur, the substance should be considered as "poorly water soluble", not as "highly insoluble in water", and therefore long-term testing has to be considered.
The concept of "poorly water soluble" is associated with the need to consider long-term tests instead of short-term tests. The ECHA Guidance on Information Requirements and Chemical Safety Assessment section R.7.8.5 (Endpoint Specific Guidance R.7.b) suggests that water solubility below 1mg/L or below the detection limit of the analytical method of the tested substance should be used for considering the substance as poorly water soluble and performing the long-term tests instead of the short-term tests:
http://echa.europa.eu/guidance-documents/guidance-on-information-requirements-and-chemical-safety-assessment
For further details regarding testing on aquatic toxicity please consult OECD Guidance Document on Aquatic Toxicity Testing of Difficult Substances and Mixtures available at:
http://www.oecd-ilibrary.org/environment/guidance-document-on-aquatic-toxicity-testing-of-difficult-substances-and-mixtures_9789264078406-en
- The scenarios that a (potential) registrant may encounter
- (potential) registrant’s obligation to submit an inquiry dossier
- the type of inquiry to be specified in section 14 of the IUCLID dossier.
| My situation | Should I submit an inquiry? | Type of inquiry |
|---|---|---|
| I am planning to register a substance | There is always an obligation to make an inquiry before registering a substance, in accordance with Article 26(1) of the REACH Regulation.This guarantees that you are put in touch with all previous registrants of the substance as well as other potential registrants. | Inquiry for a substance before its registration (Type 3) |
| I am the Lead registrant and I want to increase the tonnage band of the joint submission | Yes, an inquiry should be made in situations when a registrant reaches the next tonnage threshold (Article 12(2)). | Inquiry for tonnage band increase (Type 4) |
| I am a member of a joint submission and I want to update my registration from intermediate to full | Yes, an inquiry should be made to transition between the information requirements of Article 17(2) or 18(2) to the information requirements stemming from Article 10. | Inquiry for tonnage band increase (Type 4) |
| I am a member of a joint submission and I want to increase my tonnage band within the tonnage band covered by the joint submission but I don't have data | Yes, in accordance with Article 12(2 | Inquiry for tonnage band increase (Type 4) |
| I am a member of a joint submission and I want to increase my tonnage band within the tonnage band covered by the joint submission | If the information is available in the joint submission and there is an agreement in place on sharing additional data you might want to contact directly the lead registrant. Otherwise, submit an inquiry to ECHA. | Inquiry for tonnage band increase (Type 4) |
| I am a registrant with an individual registration, and I want to increase the tonnage band of my registration or change the registration type from “Intermediate” to “Full”. | Yes, in accordance with Article 12(2), if you need to comply with new (higher) data requirements for the update. | Inquiry for tonnage band increase (Type 4) |
| I am the Lead registrant and I want to change the registration type of the joint submission from “Intermediate” to “Full”. | Yes, in accordance with Article 12(2), if you need to comply with new (higher) data requirements for the update. | Inquiry for tonnage band increase (Type 4) |
Further details on the inquiry procedure can be found on ECHA’s website.
The Inquiry process aims to put potential registrants and previous registrants in contact with each other to share data so that the joint submission obligations can be met.
Studies involving vertebrate animals should not be repeated and available studies need to be shared. By doing so, it reduces registration costs and avoids unnecessary testing, especially on vertebrate animals.
Further details on the inquiry procedure can be found on ECHA’s website at: http://echa.europa.eu/regulations/reach/substance-registration/inquiry.
In principle, the analytical data included in an inquiry or a registration dossier must reflect the substance as manufactured or imported. Hence, if you are an EU manufacturer, you must submit analytical data generated from a sample that you have manufactured. If you import from outside the EU, you must submit analytical data generated from a sample manufactured by the non-EU manufacturer.
We are aware that this can be problematic if you have to register your substance before taking up manufacture or import. For such cases we may accept submitting analytical data from another source.
For these exceptional cases, you need to explain the following in your inquiry dossier:
- Why the analytical data cannot be generated on the manufactured substance.
- Why the substance you intend to manufacture or import will be the same as the one used to generate the analytical data. For example, a statement that the manufacturing process and/or plant specification used to produce the analysed substance will mirror that for the inquired substance.
You also need to provide the following information in your inquiry dossier:
- The source of the analysed substance i.e. manufacturing site name and address.
- A short description of the production process and the raw materials for both the inquired substance and the analysed substance.
- The foreseen manufacturing or import volume for the inquired substance.
- A statement from the owner of the analytical data indicating that you have their permission to use their analytical data
You are required to update the substance identification information in the registration dossier within 6 months after the submission to reflect the substance as manufactured or imported. If the registration is not updated, ECHA may initiate a targeted compliance check on substance identity.
Inquiry dossier should be prepared the same way regardless it is for intermediate or for full registration. The purpose of the inquiry process is to ensure data sharing by all registrants and potential registrants of the same substance, so that the joint submission obligations are met. Thus, despite of the type of registration you plan to submit (full or intermediate) it should contain sufficient analytical information to ensure the accurate identification of the substance.
An inquiry dossier should contain:
- Information about the potential registrant.
- Information about the identity of the inquired substance. The analytical data (qualitative and quantitative data) provided in the inquiry dossier should be sufficient to verify the identity and composition of the substance.
- Information requirements where the potential registrant would need to carry out new studies, including those involving vertebrate animals.
As a result of submitting an inquiry the potential registrant is put in contact with previous registrants and other potential registrants of the substance. By doing so, we ensure that data is shared by all registrants of the same substance and that the joint submission obligations are met.
Before submitting any dossier, we recommend that you use the "Validation Assistant", which will identify the fields of your dossier which deserve particular attention. You can download it via the IUCLID 6 website https://iuclid6.echa.europa.eu/.
The Validation Assistant supports the preparation of your IUCLID 6 dossier in two ways:
- It performs a check of most of the business rules applied to dossiers in REACH-IT. This enables the user to detect and correct failures before submitting the dossier to ECHA. For further information on the business rule check, you can consult the Manual How to prepare registration and PPORD dossiers, available at: https://echa.europa.eu/manuals
- It performs a so-called "Substance Identity check", identifying the IUCLID fields of an inquiry dossier that should be filled in or that need particular attention. It is advisable to use the Validation Assistant both for preparing the inquiry substance dataset and the final dossier. We strongly recommend addressing all the reported inconsistencies and shortcomings. Please note that the "Substance Identity check" will not assess whether the information submitted is adequate but only if all required fields are filled in.
If you are uncertain about your substance’s identity, do not include any numerical identifiers (i.e. EC, CAS) in the reference substance of your inquiry dossier. Instead, write “tentative name” followed by a proposed representative chemical name in the IUPAC name field (e.g. tentative name, Reaction mass of A and B). Refer to any other relevant identifier in the "Other substance identifiers” field. See screenshot for clarity.
After receiving the appropriate identifiers for your substance as part of the inquiry outcome, make sure you update the IUPAC name field accordingly when preparing the registration dossier.

If you have unambiguously identified your substance, you should include any available numerical identifiers (i.e. EC or CAS) in the reference substance field of your inquiry dossier. After your inquiry is assessed, ECHA will grant you full access to the Co-registrants page of your substance.
If the EC number is not available to you and the substance does not have a CAS number but you are already in contact with the lead registrant, you can request the identifier from the lead. Otherwise, you may request the identifier from ECHA using the web form: http://echa.europa.eu/en/web/guest/contact
ECHA does not check the completeness of the substance identity information submitted as part of the inquiry process. The completeness check will only be performed on the registration dossier, in accordance with Article 20 (2) of the REACH Regulation.
However, you can minimise the risk of failures before you submit the registration dossier by using the IUCLID Validation assistant tool. We advise you to validate the dossier and correct the information by following the advice reported in the tool.
If the Validation assistant does not indicate any failures, it is not an automatic confirmation that your dossier is complete, since the technical completeness has been complemented with additional verifications done by ECHA staff that are not displayed in the Validation assistant report. Information on the areas of the additional verifications can be found at: https://echa.europa.eu/documents/10162/13652/manual_completeness_check_en.pdf
- Contact details: names and addresses of the previous registrants and, if appropriate, other inquirers (potential registrants).
- Studies: a list of submitter’s contact details for each endpoint and the UUIDs of the (robust) study records which have been submitted more than 12 years earlier. This information enables the potential registrant to request the sharing of existing data from the previous registrants.
- Contact details: names and addresses of the previous registrants and, if appropriate, other inquirers (potential registrants).
- Studies: a list of the submitters’ contact details for each endpoint and the UUIDs of the (robust) study summaries which have been submitted more than 12 years earlier. This information enables the potential registrant to request these robust study summaries directly from the previous registrants.
No. There is no deadline for submitting a new inquiry dossier to ECHA.
No. Before submitting your registration, you need to wait until you have received a communication from ECHA which includes the inquiry number. We will also provide you with the link to the relevant co-registrants page in REACH-IT, where you will find the details of registrants and successful inquirers of the same substance. This will support you in complying with your obligations to share data and submit a joint registration.
- a list of submitter’s contact details for each endpoint
- the UUIDs of the (robust) study summary which have been submitted more than 12 years earlier.
No. You need to wait until you have received the communication from ECHA, which states your inquiry number together with the list of the requested (robust) study summaries available to ECHA. This will then allow you to determine which further studies may need to be conducted. REACH requires that new testing of a substance involving vertebrate animals can only be carried out as a last resort.
For chemicals manufactured or imported in a quantity of 100 tonnes or more, you are not allowed to conduct any vertebrate testing for the information requirements specified in Annexes IX and X of REACH. Instead, you must submit a testing proposal in your registration dossier. We will then evaluate whether the testing proposal is adequate before allowing you to perform the test.
ECHA will not publish any substance identity information submitted as part of an inquiry.
ECHA uses the information submitted for the purposes of inquiry solely to determine whether the same substance has been previously registered or inquired about. We make available the contact details and list of information requirements only within the relevant Co-Registrants page in REACH-IT. The EC/list name, EC/list number and/or EC/list description is the only substance identity information disclosed to registrants and successful inquirers of the same substance.
Each time a new potential registrant successfully inquires about a substance or a registrant requests for additional information for a substance, an email notification is sent to all co-registrants in REACH-IT, informing them that a new member registrant is potentially entering the market. The new member will most likely only interact with the lead registrant, but for transparency reasons, all co-registrants are informed about it (see screenshot below).


The co-registrants page (CoRP) helps registrants fulfil their data sharing and joint submission obligations. It is accessible to registrants and potential registrants who have successfully inquired about a substance. It displays their contact details and, in case of inquirers who requested endpoints data, the list of requested information. Also, the role of the registrants within the joint submission is visible for all, so the lead can be easily identified and directly contacted for the purpose of data sharing negotiations.
Different information is displayed depending on their status:
- Potential registrants, within 1 year after successfully submitting their inquiry, can see all registrants and potential registrant(s). However only the leads are identified.
- Potential registrants, beyond 1 year of their successful submission and if they have not registered, they can no longer see any registrant(s) or any other potential registrant(s).
- Registrants can see the roles of all co-registrants, i.e. whether they are lead or member and can see the potential registrants.
They are not shown in the case where the notifying company did not claim a registration number for the notified substance. For such cases, only the company name and the country are displayed.
The co-registrant page shows only the contact details of those companies who did claim the registration number.
Yes. The Third Party Representative (TPR displayed on the co-registrants page is specific to the inquiry/registration dossier. Consequently, if the same TPR has been nominated in several inquiry or registration dossiers, the same contact details (of the TPR) are displayed multiple times.
It depends under which type of intermediate as described under Article 3(15) of the REACH Regulation your intermediate falls, whether you have registration obligations or not.
- Non-isolated intermediates:
For the use of a substance as a non-isolated intermediate, there are no obligations under the REACH Regulation.
- On-site isolated intermediates:
A manufacturer of on-site isolated intermediates in quantities of 1 tonne or more per year needs to register their substances (if they are not otherwise exempted from registration (see FAQ ID=30). However registrants of on-site isolated intermediates can provide reduced registration information according to Article 17(2) of the REACH Regulation if they confirm that the substance is manufactured and used under strictly controlled conditions as described under Article 17(3) of REACH.
- Transported isolated intermediates:
A manufacturer or importer of transported isolated intermediates in quantities of 1 tonne or more per year needs to register his substances if they are not otherwise exempted from registration. However, a registrant of transported isolated intermediates can provide reduced registration information according to Article 18(2) of the REACH Regulation if he confirms that he is manufacturing and/or using the substance under strictly controlled conditions and if he confirms or states that he has received confirmation from the user that the substance is used under strictly controlled conditions as described under Article 18(4) of REACH. In this case, both the registrant and the users are each liable for their own statement regarding the strictly controlled conditions.
When and how the specific provisions for the registration of intermediates under REACH can be used are described in the Guidance for intermediates: http://echa.europa.eu/guidance-documents/guidance-on-reach.
More information can be found at: http://echa.europa.eu/documents/10162/13655/pg16_intermediate_registration_en.pdf
As described in Appendix 4 of the Guidance on Intermediates (page 35)" due to the practical nature of manufacturing processes and to the fiscal attributes of manufacturing sites, one or more steps between the manufacturing of the substance (A) and its use in the manufacturing of substance (B) may be necessary to facilitate/ensure proper chemical processing in the synthesis of substance B." Therefore, necessary purification of the intermediate, which takes place after its manufacture and before the synthesis, does not prevent it from being considered an intermediate.
Article 3(15)(c) of the REACH Regulation does not require that the manufacture of the transported isolated intermediate and its synthesis is done on sites operated by the same legal entity.
However, in order to benefit from the reduced information requirements for registration dossiers submitted under Article 18 of the REACH Regulation, the registrant must ensure that the substance is handled under strictly controlled conditions throughout its lifecycle - also if it undergoes a purification. It must also be ensured that the substance is always manufactured for and consumed in or used in the synthesis of another substance.
The Guidance on Intermediates is available on the ECHA website at: http://echa.europa.eu/guidance-documents/guidance-on-reach
A non-isolated intermediate is defined as an intermediate that during synthesis is not intentionally removed (except for sampling) from the equipment in which the synthesis takes place.
For further information see the Guidance on registration:
Yes, under the conditions specified in the reasoning provided below.
Manufacturing of coke electrodes
Coal tar pitch, high temperature (CTPHT - EC number 266-028-2) and Anthracene oil (AO - EC number 292-602-7) are used in the manufacture of electrodes for applications in electrolytic processes in the aluminium industry (i.e. aluminium smelters). These substances are used specifically to manufacture the following types of electrodes:
- Søderberg electrodes – these are first manufactured directly in the electrolytic cell and subsequently used in the same cell.
- pre-baked electrodes - they are manufactured in dedicated units and later used in the electrolytic cells. Electrodes manufacturing units may be located in the same site as the electrolytic cells or in another site.
In both cases, the manufacturing process of the electrodes consists of the following stages:
- Mixing of the raw materials (so called filler grains usually petroleum coke or calcinated coke + CTPHT and/or AO)
- Shaping (to give the electrode the final shape which is required to fit it into the housing of the electrolytic cell)
- Baking
The outcome from the baking process is "Coke", a new substance. The new substance is manufactured from petroleum coke or calcinated coke, AO and CTPHT that contribute to its structure.
In more detail the baking process works as follows:
- Coke substances are carbonaceous materials obtained from coking processes such as baking at relatively high temperature. These substances are characterised by a high carbon elemental content and can display unique structures presenting a high carbon-to-hydrogen ratio. The exact composition of coke is generally complex and depends on the source used and the conditions applied for the coking. As a source of carbon, coke substances find applications in processes such as aluminium manufacturing by electrolysis. In this specific case, CTPHT and AO are themselves chemically transformed into coke during the manufacturing process of pre baked and Søderberg electrodes. These transformations involve complex chemical reactions including polycondensation and polymerisation of the constituents which CPTHT and AO consist of. These reactions begin during the baking process in a low oxygen atmosphere at temperatures of ~400 C. The transformation process into coke is completed at around ~700 C with the condensation of all polyaromatic hydrocarbons. The transformation leads to a carbonaceous material of high carbon elemental content and CTPHT and AO contribute to the structure of the coke substance intended to be manufactured. In this case, the baking process is carried out in the presence of readily available coke filler grains. The result is a homogenous coke displaying specific electrical conductivity (a required feature for the coke to be used as an electrode) and mechanical strength. In this specific case, the resulting coke would in principle not have the chemical structure that would enable its use as a source of carbon in electrolytic processes without the combined use of the AO and CPTHT as precursors and readily available coke grains.
- The outcome from the manufacturing process (i.e. the coke electrode) is, in this specific case, a substance under REACH and not an article as defined in Article 3(3) of REACH. The shape of the electrode is decided to fit it into the housing of the electrolytic cell, which can be different from case to case. Furthermore, during theuse of the electrodethe carbon from coke is consumed. The specific shape, surface and design given to the electrodes during their manufacture are therefore less relevant for its use in the aluminium production process than its chemical composition.
Regulatory analysis
According to Article 3(15) of REACH, an intermediate is a substance which is "manufactured for and consumed in or used for chemical processing in order to be transformed into another substance (…referred to as synthesis)".
Whenever a substance is used to achieve another function than its transformation into another substance (e.g. as an individual step in the production process of an article), it cannot be regarded as an intermediate. It is also recognised that, due to the practical nature of manufacturing processes and to the fiscal attributes of manufacturing sites, one or more steps between the manufacturing of a substance (A) and its use as an intermediate in the manufacturing of another substance (B) may be necessary to facilitate/ensure proper chemical processing in the synthesis of that other substance (B) (see Appendix IV of ECHA's Guidance on intermediates, December 2010).
In the current case, if all stages a, b and c are an integrated part of the coke manufacturing process installations, the use of AO and CTPHT may be considered the use as an intermediate.
However, whenever the mixing of AO, CTPHT and filler grains (stage a) is not carried out on the same site, this may indicate that the mixing step is not performed to facilitate/ensure proper chemical processing in the synthesis of the coke. In that case and AO and CTPHT cannot be regarded as intermediates.
ECHA fees and charges payable under Regulation (EC) No 1907/2006 ('the REACH Regulation') are set by the European Commission, with the agreement of the EU Member States. They are laid down in the Commission Regulation (EC) No 340/2008 of 16 April 2008 on the fees and charges payable to the European Chemicals Agency (OJ L 107, 17.4.2008, p. 6, "the REACH Fee Regulation"), as subsequently amended by the Commission Implementing Regulation (EU) No 254/2013 of 20 March 2013 (OJ L 79, 21.3.2013, p. 7), by the Commission Implementing Regulation (EU) No 211/2014 of 27 February 2014 (OJ L 67, 7.3.2014, p. 1), by the Commission Implementing Regulation (EU) 2015/864 of 4 June 2015 (OJ L 139, 5.6.2015, p. 1) and by the Commission Implementing Regulation (EU) 2018/895 of 22 June 2018 (OJ L 160, 25.6.2018, p. 1), and are subject to regular reviews.
For more information, please consult the European Commission website.
ECHA does not carry out an economic activity within the meaning of Directive 2006/112/EC on the common system of value added tax. This means that no VAT has to be paid on the fees defined in Regulation (EC) No 340/2008 ('the REACH Fee Regulation'). For the reason above, ECHA has no VAT number and ECHA's invoices are exempt from VAT. ECHA's Finnish Business ID number is 2139942-8 and it is always mentioned on ECHA's invoices. However, this number is not a VAT number.
Thus it is important to note that ECHA is unable to provide registrants a VAT or a withholding tax / tax at source residence certificate. As ECHA has no tax residence in any Member State. In Q&A 739 ECHA has published a financial identification form confirming its Finnish business ID.
For any enquiry, please contact ECHA by using the ECHA Helpdesk contact form.
The registration fee for a substance depends on the tonnage of the registration, size of the company and the type of submission. Additionally:
- Lower fees apply to joint submissions as compared to individual submissions. This does not apply in case you decide to opt-out of the joint submission;
- SMEs benefit from a reduced fee in all categories;
- An additional fee is levied for confidentiality.
No fee is required for the registration of substances if you are entitled to a fee waiver. Further information related to the fee waiver can be found under Q&A 1237.
All these provisions are specified in Articles 12(1)(a) and 74 of REACH, taking into account Recital 34 of the regulation. The fee amounts can be found in the REACH Fee Regulation (No. 340/2008, and subsequent amendments).
Further information can be found under Invoicing and Payments..
Fee amounts are specified in the Commission Implementing Regulation (EU) No 2018/895 of 22 June 2018 amending Regulation (EC) No 340/2008 on the fees and charges payable to the European Chemicals Agency ("the REACH Fee Regulation").
Information related to the chargeable confidentiality claims can be found in the manual “Dissemination and Confidentiality under the REACH Regulation”, chapter 3.5.
ECHA encourages to run the Fee calculator plugin before submitting the IUCLID dossier in REACH-IT. A potential fee mismatch will enable you to identify and correct a mistake in your dossier before the submission.

The timelines for payment of fees levied under the REACH Regulation are specified in the REACH Fee Regulation as subsequently amended by the Commission Implementing Regulation (EC) No 340/2008 and its subsequent amendments (see Q&A 716) and the precise payment due date is set out in ECHA's invoice.
Fees invoiced for initial and update registrations
In the case of fees for registrations submitted, as well as in the case of updates of a registration, the initial payment due date of the fee is set at 14 calendar days from the date on which the invoice was notified to you.
If the payment has not been made within the prescribed period (by the initial payment due date), ECHA will set a second deadline for payment. This second deadline (extended payment due date) is 60 calendar days from the initial payment due date.
If the payment is not made by the extended payment due date, the registration fails the completeness check and will be rejected.
For updates of a registration unrelated to tonnage band changes, the registrant has the possibility to request a further extension of the second deadline for fee payment before its expiry. In practice, this request for extension of the second deadline for payment applies only to:
- Fees levied for new chargeable confidentiality claims included in updates;
- Fees levied as a consequence of a legal entity change.
The request for extension should be submitted before the expiry of the second deadline via the ECHA Helpdesk using the contact form.
If the payment is not made by the deadline of the extension, the update fails the completeness check and will be rejected.
Fees invoiced (PPORD) exemptions and for requests to extend PPORD exemption.
Fees for notification of PPORD exemptions: the initial payment due date of the fee is 7 calendar days from the date on which the invoice was notified to you. Requests to extend a PPORD exemption: the initial payment due date of the fee is set at 30 calendar days from the date on which the invoice was notified to you.
If the payment has not been made within the prescribed period (by the initial payment due date), ECHA will set a second deadline. This second deadline (extended payment due date) is 60 calendar days from the initial payment due date.
Where the payment is not made by the expiry of the second deadline, the notification or the request for an extension will be rejected.
Fees invoiced for Legal Entity Change (LEC)
Fees for Legal Entity Change: the initial payment due date of the fee is 14 calendar days from the date on which the invoice was notified to you.
If the payment has not been made within the prescribed period (by the initial payment due date), ECHA will set a second deadline. This second deadline (extended payment due date) is 30 calendar days from the initial payment due date.
Where the payment is not made by the expiry of the second deadline, the Legal Entity Change will be rejected.
Fees invoiced for Applications for Authorisation (AfA)
Fees for Application for Authorisation: the initial payment due date of the fee is 14 calendar days from the date on which the invoice was notified to you.
If the payment has not been made within the prescribed period (by the initial payment due date), ECHA will set a second deadline. This second deadline (extended payment due date) is 7 calendar days from the initial payment due date.
Where the payment is not made by the expiry of the second deadline, the Application for Authorisation will be considered as not received by ECHA. In this case the application is not processed further. The only way to proceed is to re-submit the application.
ECHA does not send confirmations of receipt of payment, but you can check the status of your payment in REACH-IT. Open the submission report and look at the “Submission processing steps” section; the "Financial completeness check" step will be green when your payment has been received and accepted by ECHA.
Please note that it may take several days before ECHA receives the payment depending on the payment method. A SEPA payment is transmitted within three bank business days.
A submission is subject to both a technical and a financial completeness check. Therefore, a fully paid invoice does not necessarily indicate an accepted submission of your REACH dossier.
When an invoice is cancelled (e.g. due to incorrect information on the invoice, such as billing information, company size, etc.), you will receive a credit note for the whole amount and, if needed, a replacement invoice. These electronic documents can be downloaded from your REACH-IT account in PDF format. If you receive two invoices and one credit note, the reference number you should indicate in your bank transfer is the reference number of the latest invoice.
If you pay an invoice for which ECHA later issues a credit note, the paid amount will be credited back to your company by either reallocating the received amount to another open invoice or refunding the amount to you.
Please be informed that ECHA doesn't provide any credit notes in case the invoice was not paid in full and your registration was rejected.
- Company name and address indicated by the registrant (or the billing details, if the tab "Billing company information" was filled in by registrant);
- Customer ID number (= Legal entity UUID);
- DUNS number and VAT number (if provided by the registrant);
- Purchase order number (if provided by the registrant when submitting the dossier or when accepting a legal entity change).
If your invoice has not been paid yet, ECHA will be able to cancel it and create a new one with your updated company information. If the invoice has already been paid, ECHA cannot modify the invoice.
ECHA is unable to update any company information in REACH-IT. It is the responsibility of the REACH-IT account owner within the company to make sure that the information is up-to-date.
To receive an invoice with updated company information update your information in REACH-IT. Go to Menu >> Manage Company >> Company information. Here you can modify and update your information by clicking on "Update" at the top right corner of the page. Always remember to save your changes. Additional information on how to update your company information can be found in the ECHA Accounts Manual for Industry Users.
After the company details have been updated, you need to contact the ECHA Helpdesk by using the contact form available on the ECHA website.
Company size information
If the changes in your company information are related to a change in the company size or to an incorrectly declared company size, please refer to the SMEs section on the ECHA website.
You are entitled to the fee waiver if:
- Your substance is a phase-in substance and considered to be low risk (i.e. does not meet the Annex III criteria of REACH); and
- You submit a standard registration dossier for 1-10 tonnes per year voluntarily providing the full set of information listed in Annex VII.
The fee waiver does not apply if:
- Your substance is considered to be low risk (i.e. does not meet the Annex III criteria) and you submit a registration dossier for 1-10 tonnes per year with only physicochemical information (listed in section 7 of Annex VII); or
- Your substance is not considered to be low risk (i.e. meets the Annex III criteria) and therefore, you must provide the full Annex VII information; or
- Your substance is a non-phase-in substance.
The above is based on REACH Regulation Articles 12(1)(a) and 74, taking into account Recital 34.
Important notes
In a joint submission, the tonnage band of the other member registrants does not influence the applicability of the fee waiver for your own dossier.
You need to actively claim the fee waiver in IUCLID to benefit from it – the system does not grant it automatically.
More information on the Annex III criteria and their applicability can be found here.
When submitting a dossier or accepting a legal entity change, you are able to indicate your own internal purchase order number in the third step of your dossier creation wizard (see screenshot). The indicated purchase order will be linked to the invoice issued for the dossier submission or the legal entity change.
For the submission report or an already issued invoice, it is not possible to add any forgotten purchase order number aor modify it afterwards.

As you register online in REACH-IT you should not send any purchase orders by ordinary mail or email to ECHA. Please make sure that your system does not send any purchase orders automatically to ECHA. Your purchase order is only for your internal purposes. ECHA will not confirm your internal purchase order or provide information to or access third party platforms for handling your purchase orders.
Bank transfer is the only accepted payment method. Other types of payment, such as cheque, banker's draft or cash, are not accepted by ECHA.
Payments should be executed in Euros only and it must be ensured that the full amount of the invoice is received by ECHA. Deductions of bank charges, from exchange rate differences or of any other costs will lead to an underpayment of the invoice.
The following bank details are indicated on all invoices issued under the REACH Regulation as from 01.07.2019:
Bank: ING Belgium, Avenue Marnix 24, 1000 Brussels, Belgium IBAN: BE93 3631 8789 1767 BIC/SWIFT: BBRUBEBB
Please note that separate instructions for the payment of Appeal Fees exist. These are explained in the relevant section of the Board of Appeal on ECHA's webpage.
In the payment instruction, the invoice number should be indicated as the reference number. According to Article 17(1) of the REACH Fee Regulation (EU) No340/2008, in its latest version (see Q&A 716), every payment must indicate the invoice number. Thus, in the free text message/communications or reference field of the payment you should indicate only the payment reference indicated on the invoice. It is comprised of eight digits and you will find it next to ECHA's bank details on the invoice. This is very important, as your payment is automatically processed by REACH-IT. Please instruct your accounts payable/payments department and your bank accordingly.

Where the payment of a fee is not made by the extended payment due date or by the extension of the second deadline, the submission will be rejected or deemed not to be received. In the above mentioned cases, a rejection due to non-payment is not related to the status of the technical completeness check (TCC). Your submission will be rejected regardless of a given prolonged deadline to fulfil information requirements by submitting further information.
Furthermore, if your submission is rejected, the REACH Fee Regulation (EU) No340/2008, in its latest version (see Q&A 716), stipulates that the fees paid in relation to that submission before its rejection shall not be refunded or otherwise credited to you. After you have received the rejection letter, you are able to start your submission process from the beginning.
Since 1 November 2009, a SEPA payment must be transmitted within three banking business days. The bank statement of any given day is available to ECHA only on the next banking business day.
Therefore, it normally takes up to five banking business days before ECHA can handle your payment and confirm the invoice as paid in REACH-IT. Please note that the handling time may be longer if your payment cannot be dealt with automatically. By following ECHA's instructions regarding payments you can significantly reduce ECHA's handling time of your payments (see Q&A 721).
On the basis of the daily electronic bank statement, REACH-IT automatically matches your payment with your open invoice if you have indicated the correct payment reference (invoice) number in your payment message and you have paid the correct amount. An additional condition is that you pay each invoice separately as a single payment. One bank transaction per invoice therefore ensures the fast registration of your payment.
ECHA strongly advises you not to pay two or more invoices in the same transaction ("multiple invoice payment"). If you however include more than one invoice in the payment, please make sure that the invoice references are mentioned in full. Otherwise, the payment has to be manually handled by ECHA's accounting department. This may delay the processing of the payment and thus also the overall completeness check of your registration. If, for some reason, you cannot avoid sending multiple invoice payments and you are unable to indicate the full reference numbers in the payment, please make sure to follow the instructions in Q&A 732, which describes the sending of payment advices.
- Pay one invoice per transaction (single payment);
- Indicate the correct reference number (eight digit invoice number);
- Ensure the invoiced amount is paid in full (no charges or exchange rate differences);
- Instruct your bank to send a SEPA payment with shared cost.
All the information you need for proceeding with the payment is visible in the invoice itself. However, depending on your internal administrative procedures, it is advisable that your accounting/accounts payable department is prepared for the payment of ECHA's invoices.
We therefore recommend that you set up ECHA's bank account information in the accounting system well in advance of the first invoice's due date.
PLEASE NOTE OUR NEW BANK DETAILS SHOWN ON INVOICES AS FROM 01 JULY 2019.
On 01 July 2019 ECHA changed the bank account details shown on its fees and charges invoices. The Agency’s new house bank is ING Belgium NV. ECHA’s new financial identification form (see attachment) covers all the details needed for this new bank account.
If you use an external accounting company, please ensure that all required information about ECHA and REACH is passed on, in order to ensure the smooth handling of ECHA's invoices. It is also advisable that you ensure beforehand that there are no misunderstandings related to ECHA's status as an EU body which is exempt from any national tax or any value added tax. Please refer to Q&A 725 and Q&A 726.
Please make sure that the persons handling the payments of ECHA's invoices are aware that the payment should be made at the full amount so that any possible bank charges are not deducted from the payment.
Please inform your accounting department that if the invoice is paid after the extended due date, the REACH dossier submission will be rejected and the paid fee will not be refunded.
Please ensure that ECHA’s invoice number is stated in the payment instruction (see Q&A 721).
An overview of what must and what may be jointly submitted for registration based on Article 11 of the REACH Regulation is provided in Section 6.2- 'Overview of the part of the technical dossier that may be jointly submitted for Registration' of the Guidance on data sharing: http://echa.europa.eu/guidance-documents/guidance-on-reach.
Some information of the registration has to be submitted jointly whereas other information needs to be submitted separately. Additionally, there is information the registrant(s) may decide themselves whether to submit jointly or separately, according to the criteria defined in Article 11(3) of REACH.
The following information must be submitted jointly: information on the classification and labelling of the substance, (robust) study summaries and an indication as to which of the submitted information on classification and labelling, study summaries and robust study summaries has been reviewed by an assessor. Under specific conditions, which should be explained in the dossier, a separate submission of these data is allowed (see also Q&A 109).
Additionally, each registrant must submit individually: the identity of the manufacturer or importer, the identity of the substance, information on the manufacture and use(s), exposure information for substances in quantities of 1 to 10 tonnes and an indication of which of the submitted information on manufacture and use has been reviewed by an assessor. The registrants may decide to submit the following information jointly or separately: guidance on safe use of the substance, a Chemical Safety Report (CSR) when required and an indication which of the information submitted for the CSR has been reviewed by an assessor.
Member registrants can submit the following information separately, as specified in Article 10(a) (iv), (vi), (vii) or (ix) of REACH:
- The classification and labelling;
- Study summaries of the information derived from the application of Annexes VII to XI;
- Robust study summaries of the information derived from the application of Annexes VII to XI, if required under Annex I; and
- Proposal for testing where listed in Annexes IX to X.
Article 11(3) of REACH allows an "opt-out" under specific conditions. Such an "opt out" can only cover some or all of the endpoints submitted by the lead registrant on behalf of all member registrants. However, the member registrants have to remain part of the joint submission regardless of whether information is shared.
According to Article 29(2) of the REACH Regulation, one of the main aims of the SIEF is to agree on classification and labelling where there is a difference in the classification and labelling of the substance between potential registrants.
Nevertheless if all member registrants agree, the lead registrant may include different classifications of the substance in the joint part of the registration dossier, e.g. if different impurity profiles lead to different classifications.
In this case, member registrants should leave the pertinent section of their member dossier empty to avoid being treated as an opt-out for the classification and labelling of the substance.
If the member registrants cannot agree on the inclusion of all the different classifications of the substance in the joint part of the registration dossier, one or more of the member registrants may decide to provide their substance classification separately (by filling in the respective section in their member dossier). If this is the case, a justification in accordance with Article 11(3) of REACH is required. In addition, in cases where a harmonised C&L for a substance is provided in Annex VI to the CLP Regulation, then that harmonised C&L must be used.
Further information can be found in the manual ‘How to prepare registration and PPORD dossiers’
Yes. According to Annex VI to the REACH Regulation any physicochemical, toxicological and ecotoxicological information that is available and relevant must be provided in the registration dossier.
In practice, after gathering and assessing all existing information, the registrant has to select the information that is reliable, relevant and adequate.
For key studies, robust study summaries have to be provided; for supporting studies, study summaries are sufficient.
Further guidance on information gathering and evaluation is also provided in chapters R.3 and R.4 of the Guidance on information requirements and chemical safety assessment.
According to Article 11(1) of the REACH Regulation, the information specified in Article 10(a)(ii), i.e. details on the substance identity including spectral data and chromatograms, have to be submitted separately by each member registrant of a joint submission.
This information is necessary for ECHA to be able to check the sameness of the substance submitted by the different member registrants. Therefore, generic spectral data or chromatograms must not be used. Each member registrant of a joint submission has to provide the specific spectral data and chromatograms for the substance they intend to register.
The REACH Regulation does not prevent the change of a lead registrant.
It is up to SIEF participants to agree on who is the lead registrant, who acts with the assent of the other registrants for the same substance. The SIEF/co-registrants can agree to transfer the lead role to another registrant at any point.
Information on how to transfer the lead role in REACH-IT can be found in Q&A 0380.
Multiple registrants of the same substance share two main obligations under the REACH regulation: data sharing and joint submission obligations. Registrants can identify who else has registered their substance and therefore shares common obligations under REACH.
It is the common responsibility of all (potential) registrants, yourself included, to form a single joint submission. ECHA strongly recommends that all registrants use this new page as a tool to ensure compliance with these obligations. For example, no role is displayed next to a registrant that has submitted a registration dossier outside of an existing joint submission. They are required to contact the lead registrant, as they share the same responsibility as the other multiple registrants. Registrants of the same substance are obligated to make every effort and to ensure that they are part of the same joint registration dossier. Existing registrants outside the joint submission are required to negotiate access to the joint submission with their co-registrants – regardless whether they need to share data or not.
For further information see Guidance Chapter "6 Registration: Joint Submission" Please also note the partial exceptions applicable for intermediate registrants.
To delete a joint submission search for your joint submission in REACH-IT and use the ‘Delete joint submission’ functionality.

You can only delete the joint submission if:
- There are no members in the joint submission (active or inactive)
- There are no submissions linked to it (failed or passed)
If the above options do not apply, ECHA can delete the joint submission upon your request. In order to do so, please contact ECHA using the contact form.
Please take into account that once the deletion has taken place, ECHA will not be able to revert the action.
The joint submission is created by the lead registrant using the pre-registration number, inquiry number or the registration number.
When none of these identifiers are available, or the pre-registration number refers to a submission that does not correctly define the substance identity, you should proceed as follows: Select Menu >> joint submission >> Create new.
Read carefully the information provided on the right side of the page. If you are certain that you do not have any reference number, click on the link ‘substance identity manually’ and follow the wizard.
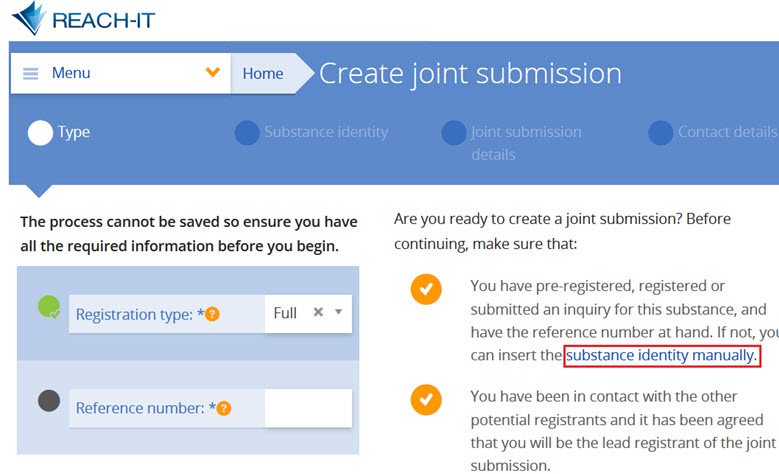
When forming a joint submission, you are required to first agree on the substance’s chemical name with all of your co-registrants. Therefore, we recommend that you first read ECHA's "Guidance for identification and naming of substance under REACH and CLP" chapter 4.3. UVCB substances. Also consult our Sector-specific support for substance identification, on certain UVCB substance types (e.g. oleochemicals, essential oils, metals, etc.).
The chemical name of the UVCB substance must be reported in the ‘IUPAC name’ field of the reference substance in section 1.1. of your IUCLID dossier. This applies even if the naming conventions for UVCB substances do not follow the IUPAC nomenclature.
The spelling of the chemical name is important, the name needs to be identical both in your joint submission in REACH-IT and in IUCLID dossier section 1.1 ‘Identification’. This is important in order to pass the business rules check. The name should not contain spelling errors or additional spaces between words or characters.
Creating a joint submission
- Start your registration by creating a new joint submission. Use the ‘insert the substance identity manually’ link, found in the checklist of joint submission creation wizard (see picture).
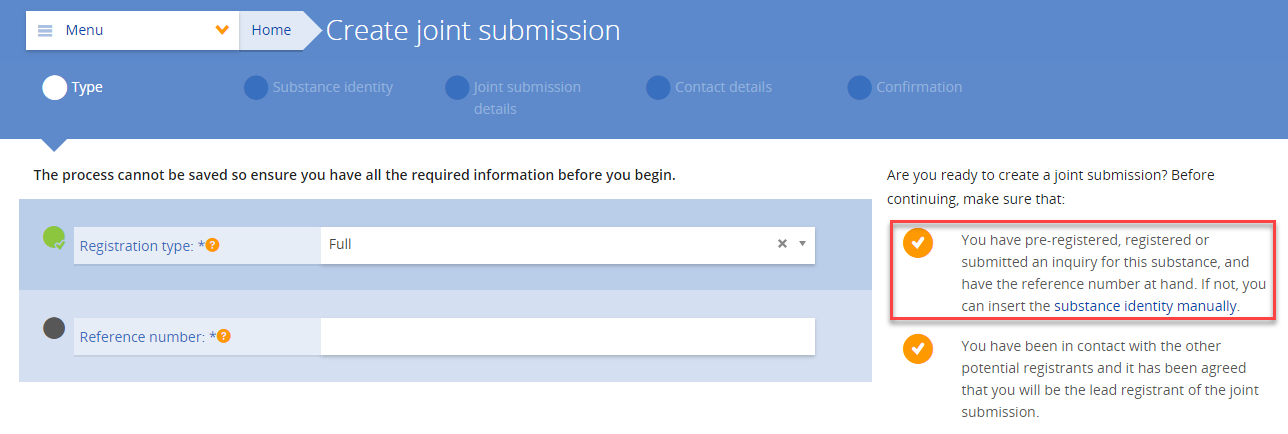
- In the next step, search for your substance name and then create the joint submission by its ‘chemical name’ (see picture).
- Select ‘other name’ or ‘IUPAC name’ depending on the naming convention of your substance as the ‘chemical name type’ and insert the derived name of your UVCB substance into the field and click continue (see picture). This will be the name of your substance in REACH-IT.

- REACH-IT might suggest other already pre-registered or registered constituents with similar names; please ensure that you select ‘your constituent’ at the bottom of the page and click ‘continue’ (see picture).

- The name has now been added to the section ‘selected constituents’ (see picture). Click ‘continue’ and confirm you substance identity and continue with the joint submission creation wizard.

Prepare and submit your dossier as part of the joint submission you created.
Remember to report all the identified constituents of your UVCB substance in IUCLID dossier section 1.2 ‘composition’. For further information on composition requirements for UVCB substances consult our guidance “How to prepare registration and PPORD dossiers.” on page 32. Also, fill in the ‘Description’ in section 1.2 including the identity of the starting materials and a description of the production process used to manufacture the substance. For Manufacturing process description see Q&A 1199.
Once you have submitted your dossier
A numerical identifier will be issued to your substance once it has passed the business rules check. Export the EC number from the joint submission page and use it in the reference substance in any future updates. Members submitting after the lead has passed the business rules check should export the issued EC number from the joint submission page and use the numerical identifier and the correct IUPAC name in section 1.1 of the IUCLID dossier. Instructions on how to download and export the EC number can be found in Q&A 1258.
Joint submissions have a status of either active or closed. Registrations can only be submitted to an active joint submission. Upon creation, the joint submission will automatically have the status active. Within the joint submission, a deadline to submit a lead dossier that at least passes the business rules verification step is clearly displayed. If no submission takes place until the given deadline, the joint submission’s status will turn to closed.
Closed joint submissions will remain in the system, but no submissions can be made into them. A closed joint submission will not block the creation of another joint submission for the same substance and same scope (full or intermediate). Therefore, once a joint submission’s status has been changed to closed, any (potential) registrant can create a new joint submission for the same substance.
Closed joint submissions cannot be turned back to active. If the joint submission is still needed, the lead registrant can create a new joint submission, using the same substance identifiers. However, a new name will be required, as joint submission names cannot be recycled.
If the current lead registrant does not want to continue in their role as lead, it is up to the SIEF members to choose a new member, who will take over the lead role. In order to transfer the role, the following shall be observed:
- The current and the future lead registrant need to agree on the handover. Then, the following actions must be performed within the Joint Submission in REACH-IT:
- The current lead registrant will need to assign the lead role to the future lead registrant using the ‘Assign New Lead’ button. The future lead registrant, should already be a member of the joint submission.
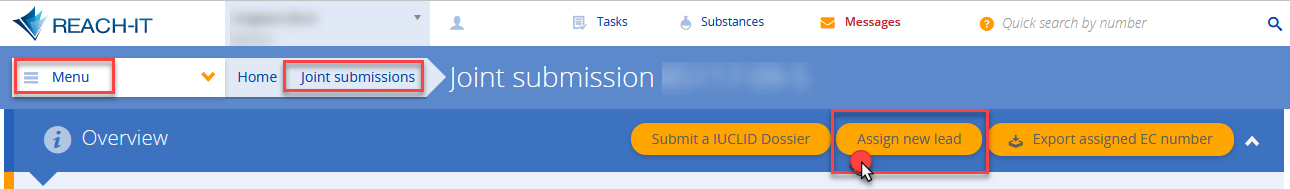
- Then, the future lead registrant should accept the assignment by clicking ‘Confirm Lead’ button and subsequently become the lead registrant in the Joint Submission.
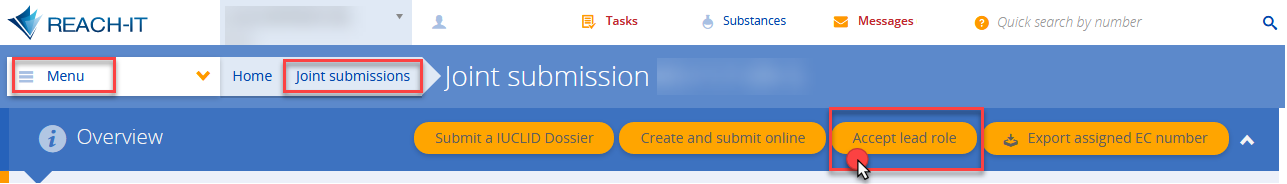
- The current lead registrant will need to assign the lead role to the future lead registrant using the ‘Assign New Lead’ button. The future lead registrant, should already be a member of the joint submission.
- The new lead registrant needs to submit a lead dossier containing all the information to be provided on behalf of the joint submission. If the new lead registrant has previously submitted a registration dossier for this substance, the lead dossier should be submitted as a spontaneous update.
For the time being, any active registration that is part of a joint submission can be updated without any limitations, as long as you are not changing the type of your registration. For intermediate joint submissions, changing your registration to both intermediate and full or only full is only possible, if there is no full joint submission already created for the substance, or there are no full registrations outside of a joint submission.
However, we highly encourage you to start negotiating with the other joint submission on merging into one, as in the future it will be unavoidable. For further information see Q&A 1170.
To increase the Joint Submission tonnage band, the Lead Registrant needs to submit a spontaneous update. Please note, that the increase of tonnage band of the Joint Submission may require additional data to be provided in the Lead Dossier.
In Dossier Creation Wizard
Step 1
The tonnage band indicated in submission type (IUCLID 6 template) represents the tonnage band of the Joint Submission. Please pick the same submission type as was used from the last successful submission at a higher tonnage band.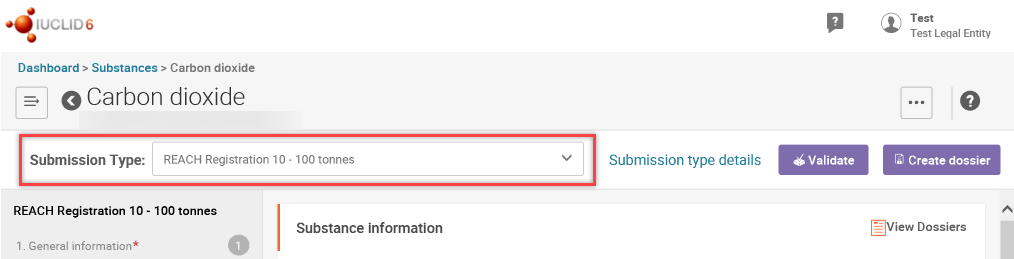
Step 2
The administrative information of the dossier represents the data of the Lead Registrant. The tonnage band indicated in this step represents the tonnage band of the Lead Registrant’s own registration.
Keep in mind that the lead registrant can have his tonnage band different from the tonnage band of the Joint Submission, while the information required for the lead dossier is based on the highest tonnage band of any Joint Submission member. If the Lead Registrant increases his own registration’s tonnage band, a new invoice will be triggered.
Mark the dossier as an update, add the last successful submission number and chose ‘change of tonnage band’ from the drop-down menu as justification for the spontaneous update.
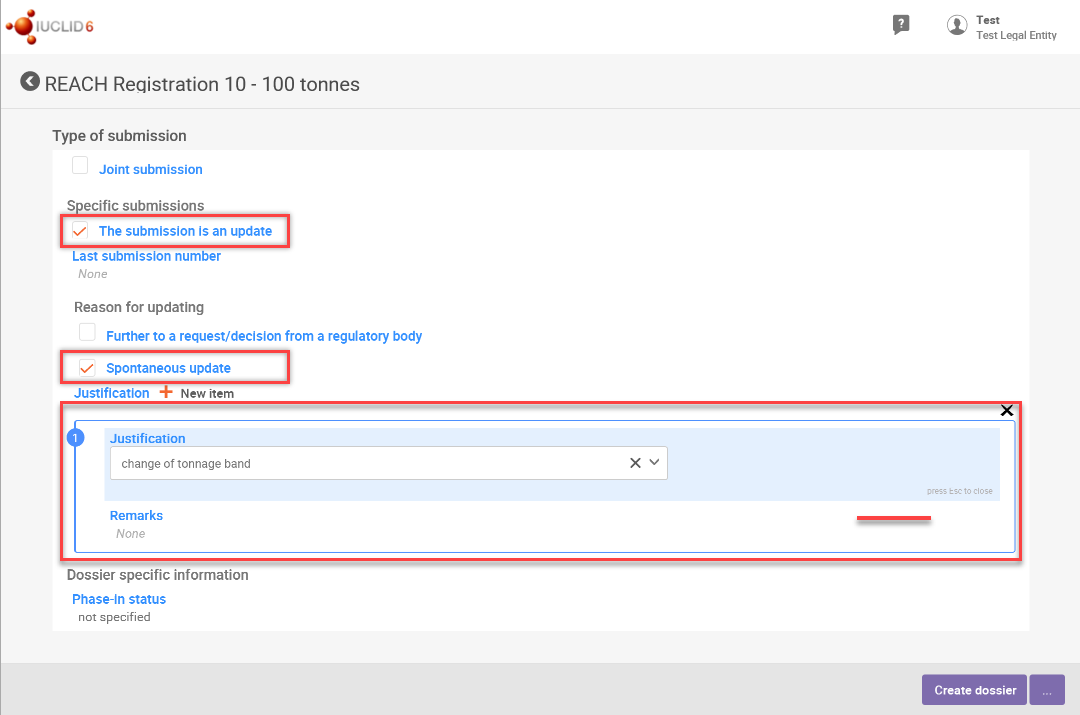
Step 1
Before decreasing the Joint Submission tonnage band, the Lead Registrant needs to make sure that all members of the Joint Submission have the tonnage band equal or below the decreased (new) Joint Submission’s tonnage band.
The tonnage band of intermediate registrations do not affect the tonnage band of the full joint submission, therefore, a member with an intermediate only registration can have a higher tonnage band than the decreased Joint Submission.
Step 2
The Lead Registrant submits an updated dossier using the same submission type standard registration or intermediate registration that was used for the last successful submission, but at a decreased tonnage band.
If the Lead Registrant’s tonnage band is above the new Joint Submission’s tonnage band, the Lead Registrant has to update its own registration's tonnage band (administrative information) to be equal or lower than the decreaed tonnage band.

Mark the dossier as an update, add the last successful submission number and chose ‘change of joint submission tonnage band’ from the drop-down menu as justification for the spontaneous update.

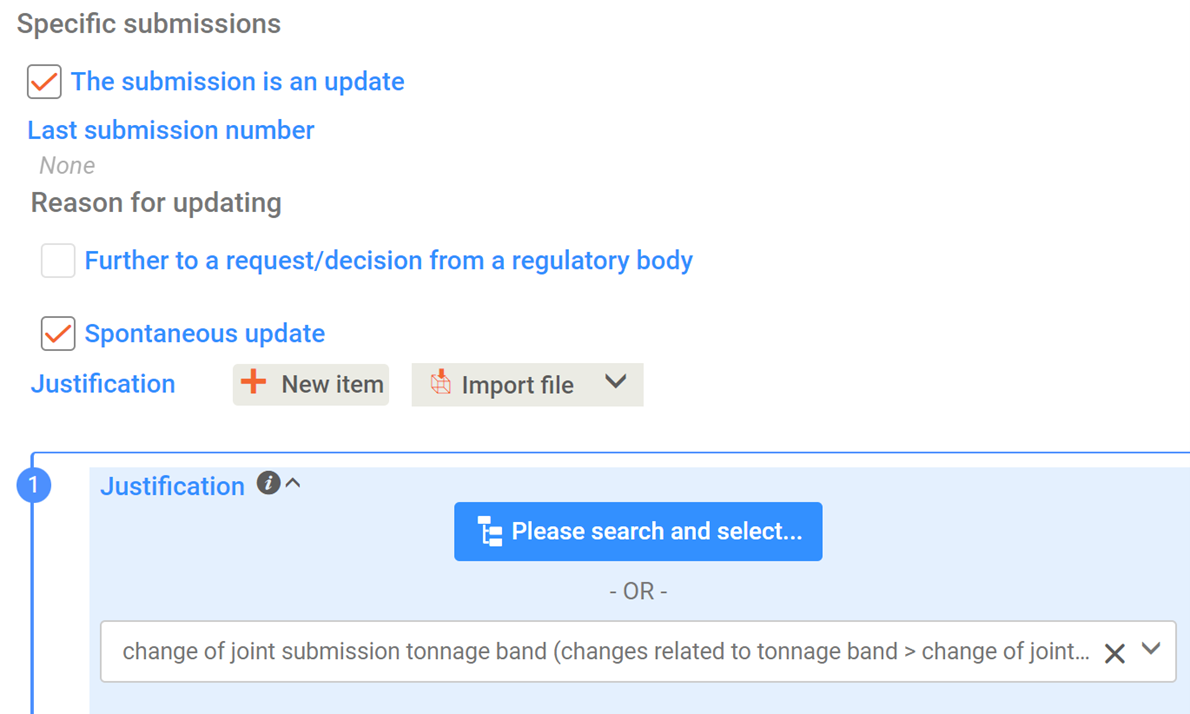
Please note: If a final decision from ECHA was sent to the Joint Submission members, they will need to comply with such decision even after the tonnage band decrease.
Yes. If the lead registrant wants to lower his tonnage band below the tonnage band of the Joint Submission, the Lead needs to submit a spontaneous update.
In Dossier Creation Wizard
Step 1.
The tonnage band indicated in submission type (IUCLID template) represents the tonnage band of the Joint Submission and should remain the same as used during the last successful submission.
Step 2.
The administrative information of the dossier represents the data of the Lead Registrant. The tonnage band indicated in this step represents the tonnage band of the Lead Registrant’s own registration, which can be equal or lower than the tonnage band of the Joint Submission, indicated in submission type.
If there is full agreement among the members of a Joint Submission and there is no Joint Submission of the different type, the lead registrant can send a formal request to ECHA so that ECHA can apply the necessary changes.
N.B. A Joint Submission type can change from full to intermediate only if there are no full members present.
Registrants of the same substance are required to register the substance jointly regardless of the use (e.g. intermediate and non-intermediate). However, due to the reduced information requirements applicable to intermediates (used under strictly controlled conditions), registrants of intermediates may choose for practical reasons to either form a joint submission together with the registrants of non-intermediate (full) or to form one parallel joint submission for intermediate use only.
Before changing your registration, please consider the following flowchart:

Please note that since registrations for: i) full, ii) transported intermediate and iii) an on-site intermediate are each considered as a separate registration and have separate fees, this update may trigger an invoice.
Data sharing negotiations.
In addition to the applicable administrative expenses, all members of a joint submission (including the lead) have to contribute to the costs of the data they need. If only one member needs additional studies, they alone will need to pay for the cost of that data. Consequently, if a registrant is planning to submit a full opt-out dossier, they are only required to share the costs related to the administration expenses of the joint submission.
For further information on how to transfer the lead role to the member of a joint submission see Q&A 380.
The lead registrant of a joint submission will provide you with a combination of joint submission name and security token outside of the REACH-IT environment (e.g. by email, phone). To confirm your membership, log in to REACH-IT and use the ‘Join existing Joint Submission’ functionality and follow the wizard.
Member registrants will also need to indicate their membership to a joint submission by adding the joint submission name during the submission of their registration dossier in REACH-IT.
Member registrants are able to indicate their membership to a joint submission in section 1.5 of IUCLID 6 registration dossier. This information can be used for your own administrative purposes, but will not be verified against the information derived from REACH-IT.
Member registrants cannot submit their dossiers until the lead dossier has been accepted for processing by ECHA. The status of the lead dossier can be checked in the joint submission in REACH-IT.
An individual registrant who wants to become part of a joint submission needs to submit a spontaneous update and indicate that their registration is part of a joint submission. Before submitting, they have to confirm membership of the joint submission, in this case the lead registrant needs to provide the joint submission name and the security token.
Once the information required is obtained, they can confirm the membership by signing in to REACH-IT >> Select Menu >> Joint Submission >> Join existing and enter the joint submission name and security token and follow the wizard.
When updating the dossier in IUCLID pay attention to the following:
- Enter the registration number in section 1.3 of your substance dataset
- Create the registration dossier by right-clicking the substance and selecting "create dossier" and follow the wizard
- Choose the template "REACH Registration member of a joint submission –general case/ - intermediate" depending on your submission.
- Indicate the reason for update as "Change from individual to joint submission"
If you are an individual registrant and no other company has registered the same substance, you will not face any constraints when updating your registration. If there are other registrants for the same substance, you will have to fulfil certain criteria to be able to update. Below you can find the scenarios of your case:
- A joint submission exists for the same substance and same registration type and your registration is outside of this: You cannot update your dossier, until you join the joint submission.
- A joint submission exists for the same substance and same registration type and your registration is part of it: You can update your registration without any limitation, as long as you are not changing the type of your registration. For intermediate joint submissions, changing your registration to both intermediate and full or only full is only possible if there is no full joint submission already created for the substance, or there are no full registrations outside of a joint submission.
- No joint submissions exist for the same substance and for the same registration type (full or intermediate), but other individual registrations have been submitted: You can update your registration without any limitation, as long as you are not changing the type of your registration or until a joint submission is created. For intermediate registrations, changing your registration to both intermediate and full or only full is only possible if there is no full joint submission already created for the substance, or there are no full registrations outside of a joint submission
As long as your registration fulfils the criteria listed below, you are allowed to update it, even if there are other individual registrations in the system:
- There are only individual registrants and no joint submission exists in REACH-IT.
- You are not changing your registration type with your update. For intermediate registrations, changing your registration to both intermediate and full or only full is only possible if there is no other full and intermediate registrant (either as part of a joint submission or as individual registrant).
If any potential or existing registrant creates and submits a joint registration, this joint registration will block all the individual registrants of the same substance and same registration type from being able to update their dossiers, until they join the joint submission.
- If there is a joint submission for the same substance you will need join the joint submission in order to submit the update.
- If there is no joint submission, but there are individual registrants for the same substance, you should create a joint submission and take up the role as lead registrant before submitting the update.
- If there is no joint submission and you are the only individual registrant, you can update your registration without creating the joint submission.
- None of the above applies, if the update requested is linked to a technical completeness check (TCC) failure. In this case you should submit an update as individual registrant.
If a potential registrant has inquired about a substance, they can find the Joint Submission and the Lead Registrant’s details by using the Joint Submission functionality and by ticking the option ‘Show other joint submissions’. See the example screenshot below and watch the explanation in this YouTube video.

If a potential registrant has not inquired about a substance, and the Lead registrant accepts to publish their contact information, they will be able to find the name of the Lead registrant on our website. The List of the lead registrants can be downloaded at the bottom of the info graphic.
If a potential registrant has not inquired about a substance and the Lead registrant does not accept to publish their contact information, they can only find the Joint Submission and the Lead Registrant’s details if:
- They submit an inquiry to ECHA. Instructions on how to submit an inquiry can be found in the manual ‘How to prepare an inquiry dossier’.
- They contact other registrants of the same substance. The contact information can be found by searching the dissemination page.
Here you can find further information on how to find your co-registrants.
- The member has to communicate their intention to upgrade their tonnage band to the lead registrant.
- The lead registrant submits a dossier update using the IUCLID template corresponding to the higher tonnage band (submission type). The dossier will include all data required for the higher tonnage band. When creating the dossier, in the dossier header, the lead registrant should indicate their own tonnage band, which will in this case be lower than the used template (administrative information).
- Once the lead has successfully submitted the lead dossier, the member can submit their updated member dossier indicating the new, higher tonnage band in the dossier header (administrative information) and pay the corresponding fee.
Fees
There is no cost linked to the increase of the joint submission tonnage band, fees are levied only for the increase of the tonnage band of a registration. Therefore, as long as the lead registrant only increases the tonnage band of the joint submission, but not their own registration, they will not incur any additional fee. On the other hand, when the member registrant submits their updated dossier at an increased tonnage band, they will receive a new invoice.
Member registrants of a joint submission (including the lead registrant) contribute only to the cost of the data they need and the applicable administrative charges. If only one member registrant needs additional studies, they alone will need to pay for the cost of that data. Should later on another registrant require the new, higher tonnage band, then the costs of the data / studies performed need to be shared amongst the registrants who require those.
Opt-out
In case the data that is required for the higher tonnage band is only available for the specific member registrant, who wishes to update their dossier, they can decide not to share it with the lead registrant, but instead include it in their own dossier and submit an opt-out for the higher tonnage band.
By submitting an opt-out dossier you will not be entitled to the reduced joint submission fees. Furthermore, any registrant who in the future wishes to update their registration to this higher tonnage band would have to include the data in their dossier separately. For this reason, ECHA recommends that the lead registrant’s dossier contains all information, regardless of their tonnage band. Alternatively, and only if the majority of co-registrants support it, the member registrant can take over the lead role of the joint submission.
Registrants of the same substance are required to register the substance jointly regardless of the use (e.g. intermediate and non-intermediate). However, due to the reduced information requirements applicable to intermediates (used under strictly controlled conditions), registrants of intermediates may choose for practical reasons to either form a joint submission together with the registrants of non-intermediate (full) or to form one parallel joint submission for intermediate use only.
Before changing the member registration, please consider the following flowchart:

Please note that since registrations for: i) full, ii) transported intermediate and iii) an on-site intermediate are each considered as a separate registration and have separate fees, this update may trigger an invoice.
Data sharing negotiations
In addition to the applicable administrative expenses, all members of a joint submission (including the lead) have to contribute to the costs of the data they need. If only one member needs additional studies, they alone will need to pay for the cost of that data. Consequently, if a registrant is planning to submit a full opt-out dossier, they are only required to share the costs related to the administration expenses of the joint submission.
You should appoint a new lead registrant together with the other registrants of the substance as soon as possible. Of course, you can offer to become the lead registrant.
A lead registrant is a legal requirement under Article 11 REACH. Moreover, there are important practical reasons why you should have a lead registrant. As such, you have an own interest in ensuring that there is a lead registrant to your registration.
For example, a lead registrant is needed to give potential registrants access to the joint submission and to confirm that they may refer to the information in the lead dossier. A lead registrant is also a natural first contact point for data sharing negotiations, and thus facilitates all registrants’ compliance with their data sharing obligations. In addition, a lead registrant is needed to update the information that was jointly submitted for a substance. Without a lead registrant, each registrant must update its registration individually with new information about the substance.
After the lead registrant has disappeared, the remaining registrants need to discuss among each other to appoint a new lead registrant as soon as possible. Once you have agreed on a new lead registrant, you should contact ECHA via the contact form, and request the transfer of the lead registrant role.
If you know that your lead registrant for a substance will disappear, you and the other registrants are encouraged to appoint a new lead registrant before the disappearance. Like that you ensure that there is no time without a lead registrant. This is easier to do in practice, because the old lead registrant can hand over the lead registrant role within REACH-IT. Q&A 0380 explains what to do step-by-step.

Registrants must always submit their information within the framework of a joint submission. However, registrants can submit some information separately, if:
- it would be disproportionately costly for them to submit some information jointly, or
- submitting the information jointly would lead to the disclosure of commercially sensitive information that would likely cause substantial commercial detriment, or
- they disagree with the lead registrant on the selection of information.
Registrants who fall under any of the above circumstances can submit some or all data on their own: these are respectively called partial and full opt-out. Technical implementation of the opt-out is described in Q&A 396.
A justification to explain why the information can be submitted separately in accordance with Article 11(3) or 19(2) of the REACH Regulation must be provided in the 'Justification' field of the record 'Opt-out information for REACH registration' in IUCLID section 14 'Information requirements'. To help you provide a valid justification there are templates available in the 'Justification' field (click the icon A). The template questions are also available in the manual ‘How to prepare registration and PPORD dossiers’, Annex 7.
Information expected in the justification:
- Cost of accessing the jointly submitted data. You should have received this information from the lead registrant. If you have not been able to obtain this information from the lead registrant you should explain the actions you took to try and obtain this information and the lead registrant’s response.
- Cost of completing and submitting your opt-out dossier. Bear in mind your own possible internal costs related to preparing the dossier. You should not provide a figure of zero for this answer.
- Explain why submitting this data jointly would be disproportionately costly. See below some examples for the basis of your explanation:
- Your registration type being different from the joint registration.
- Your registration tonnage band and the associated data requirements being different, e.g., Annex VII requirement fulfilled with an Annex VIII study.
- Type of data, e.g., data publicly available, own data available, other data available at lower cost (e.g. QSAR).
- Cost breakdown quality and clarity.
- Explain the measures taken to agree on the cost of accessing the jointly submitted data.
A justification to explain why the information can be submitted separately in accordance with Article 11(3) or 19(2) of the REACH Regulation must be provided in the 'Justification' field of the record 'Opt-out information for REACH registration' in IUCLID section 14 'Information requirements'. To help you provide a valid justification there are templates available in the 'Justification' field (click the icon A). The template questions are also available in the manual ‘How to prepare registration and PPORD dossiers’, Annex 7.
Information expected in the justification:
- You need to specify precisely which information is commercially sensitive.
- You need to explain why you believe submitting this information jointly would cause substantial commercial detriment. You explanation should show your reasoning for believing substantial commercial detriment would be caused.
- You need to explain why you disagree on the data selected. Question 1 of template ‘c’ includes a list of elements that can be relevant to this explanation.
- You need to describe what actions you had taken to try and add your data to the lead dossier and why it had not been possible to reach an agreement to add this data.
The opt-out justification requirement also applies when opting-out only for classification and labelling (C&L) information. A justification to explain why the information can be submitted separately in accordance with Article 11(3) or 19(2) of the REACH Regulation must be provided. This justification must be included in the 'Justification' field of the endpoint 'Opt-out information for REACH registration' in IUCLID section 14 'Information requirements'. For further information on how to provide a valid justification see Q&A 1722.
Before opting-out for C&L information, you should bear in mind that this information can be jointly submitted in the lead registration dossier even if it differs from the C&L of the other registrants in the joint submission. In this case, the lead registration dossier should have a separate C&L record in section 2.1 with a link to a relevant ‘boundary composition of the substance’ in section 1.2. If you decide to opt-out for the C&L information, you should therefore explain in your justification why an agreement to include the information in the lead registration dossier could not be reached. For further information on how to report the boundary composition you can consult the Q&As on Substance identity profile.
Endpoint summaries are not considered as opt-out information but are still required if you are opting-out with endpoint information or are providing your own Chemical Safety Report.
When opting-out, some of the sections that are mandatory for a complete dossier are not automatically pre-selected for inclusion in the IUCLID dossier when you use the joint submission member template. Therefore, when creating your dossier endpoint summary records including section 6 ‘Ecotoxicological information’ and section 7 ‘Toxicological information’ must be selected manually. Guidance on how to include the endpoint summaries in the opt-out dossier can be found in the manual ‘How to prepare registration and PPORD dossiers’, section 9.2 How to include endpoint summaries in an opt-out dossier.
- The reason for the disagreement with the lead registrant on the classification and labelling information. See below some examples for the basis of your argumentation:
- Quality (reliability, adequacy) of data used in deriving the classification
- Difference in interpretation of data for classification
- Difference in concentrations of constituents / impurities / additives influencing classification
- Difference in (nano)forms of the substance influencing classification
- An explanation of the differences in the classification and labelling information that you wish to submit separately compared to the jointly submitted information. For each different classification, provide a reference to the data underlying the classification. This reference may be to information submitted in your own dossier as a (robust) study summary, or alternatively, references to publicly available literature may be provided.
- An explanation of the actions you have taken to include your classification and labelling in the Lead dossier and your reasoning as to why it was not possible to include this information.
The company size can be updated in REACH-IT via the ‘Company size’ functionality from the main menu and selecting the ‘Update company size’ functionality.
This will open a wizard that will guide you through the process of updating the company size. Read carefully the instructions and have the documentary evidence at hand to substantiate the companies involved in the ownership structure.
Note: By declaring that your company is an SME, you become eligible to an SME fee reduction. ECHA routinely verifies the SME status of legal entities who self-identify as SMEs. We recommend that you keep your company size information up-to-date in your REACH-IT account.
The final decision on what documentation is considered adequate evidence of a ‘sale of assets’ is determined on a ‘case by case' basis in accordance with the relevant national principles of private and corporate law of the Member State where the entity is established. For this reason ECHA Practical Guide 8 – How to report changes in identity of legal entities limits itself to the general example of the asset sales agreement and does not provide further information in this regard.
The relevant documents should be presented to the national enforcement authorities upon request. Ultimately, the REACH duty holders bear responsibility for the submission of adequate documentary evidence. To ensure that it complies with the national legal order they may seek legal advice from in-house or private practice lawyers, for example.
More information on changes in identity of legal entities can be found in the aforementioned Practical Guide 8 – available at http://echa.europa.eu/practical-guides
Transferred items (i.e. registrations/notifications)
If a registration/notification for a substance is transferred from the legal initiator to the legal successor, who already has a valid registration/notification for the same substance, the status of the transferred registration/notification will be marked ‘Annulled’, because a legal entity can only have one registration for the same substance.
Tonnage band update
For transferred registrations when the originating legal entity has registered the same substance as the legal successor and the tonnage of the originating legal entity is higher than the registered substance of the legal successor, the system will keep track of the legal successor's right to a higher tonnage band. The legal successor will need to submit a spontaneous update to indicate the higher tonnage band. No fee applies for this update.
Transfer of joint submission roles linked to Registrations
Where the initiating legal entity has submitted registrations as part of a joint submission, the joint submission role (e.g. Member or Lead registrant) are transferred together with the registrations to the legal successor. If no registrations were submitted, the roles are not transferred. The legal successor need to confirm their membership to the joint submission.
Invoicing
The Agency will issue an invoice for the legal entity change pursuant to Commission Regulation (EC) No 340/2008 Article 5 (Fee regulation) if the item list contains one or more payable items.
- Items that trigger a fee during a legal entity change are: Registrations, Registration of on-site isolated intermediate, Registration of transported isolated intermediate.
- Items that do not trigger a fee during a legal entity change are: Pre-registrations, Product and Process Oriented Research and Development (PPORD) notifications, Classification and Labelling (C&L) notifications, Inquiry notifications.
Where an invoice is needed (i.e. the legal entity change includes at least one payable item), only one invoice will be issued per legal entity change. Invoices are issued to the legal successor. The company size of the legal successor after the change of legal personality will determine the fee. Pursuant to Commission Regulation (EC) No 340/2008 Annex III Table 3, a reduced fee will be invoiced to SMEs. After the invoice is paid in full, the transfer of items will take place and the legal entity change be completed in REACH-IT. If the invoice is not paid within the extended due date the legal entity change is deleted from the system and items will not be transferred. Information about the fee regulation can be found here.
For more information, see the guide How to report changes in identity under REACH and CLP.
When a legal entity change (LEC) takes place, a new submission number is created to keep track of the asset transfer. These submission numbers are assigned for administrative purposes and are not linked to a dossier.
1. Go to REACH-IT Menu > select ‘Reference numbers’

2. Select the reference number linked to the dossier you wish to download
3. Scroll to the middle/bottom of the page, and open the tab ‘Reference number history’
4. Select the last successful submission number (initial submission or submission update in the Event column) that took place before the legal entity change.

5. Click on request IUCLID file

Yes, according to Article 12 of the Fee Regulation (EC) No 340/2008 and the references therein to the definitions concerning micro, small and medium-sized enterprises as set out in the Annex to the European Commission Recommendation 2003/361/EC, both EEA and non-EEA companies are to be considered for the classification of partner and linked enterprises during the determination of the SME status of a company. Moreover, the non-EEA company is the very entity under consideration, in case when a submission is made by an Only Representative, as specified in Article 12 of the Fee Regulation.
Under REACH, registrants and applicants can benefit from reduced fees established for micro, small and medium enterprises (SMEs).
Companies declare their SME size at the time of dossier submission, based on which ECHA issues an invoice. At a later stage, ECHA carries out an ex-post assessment, to verify the correctness of the company size. The SME verification process is important to ensure that only genuine SMEs benefit from the reduced fees and that correct fees are collected.
Before declaring the SME status in REACH-IT, companies should be familiar with the rules set by the Commission Recommendation 2003/361/EC on SME definition.
Companies should upload documentary evidence to their REACH-IT account to support SME status claims. This is only required for those submissions, which are related to SME fee reductions.
The documentary evidence should include:
- Information about the ownership structure (upstream, downstream) at the time of the submission.
- The financial statements (consolidated, if available) together with accompanying notes for the two latest accounting periods before the time of the submission.
- Official certificate/information confirming the average number of employees for each of the two latest accounting periods before the time of submission.
The documents listed above shall be provided for all partner and linked enterprises, as defined by Article 3 of the aforementioned Commission Recommendation.
In case of Only Representatives, the assessment on whether the SME reduction applies is done by reference to the headcount, turnover and balance sheet information of the non-EU company represented and its linked and partner enterprises.
All documents uploaded to REACH-IT are treated confidentially by ECHA and used exclusively for size verification purposes. For transparency and traceability reasons, documents cannot be deleted. It is, however, possible to add supplementary documentation to your REACH-IT account at a later stage. The documentation is not assessed immediately, but only once the size verification process has been initiated.
The list of documentary evidence accepted by ECHA can be found in SME verification -page.
Instructions on how to upload documents and other useful information about REACH-IT can be found at Discover REACH-IT -guide.
More information on company’s size assessment and links to the relevant legislation can be found at SME fees under REACH and CLP -page
If the original version of any document is not in one of the official languages of the European Union, a certified translation should be provided.
Legal basis for certified translations is Article 13(3) of the amended Regulation (EC) No 340/2008 on the fees and charges payable to the European Chemicals Agency pursuant to Regulation (EC) No 1907/2006 of the European Parliament and of the Council on the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH). Please see it at:
http://eurlex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2013:079:0007:0018:EN:PDF
A certified translation is the one which is accompanied by a signed statement that the translator is competent in the source and target languages (a sworn translator) and that the translation is an exact and accurate translation of the source document. The certified translator should have a certificate or a stamp that identifies the services provider and his qualification.
The SME verification process relates to your information obligations at the time of submitting your registration, which includes the obligation to inform ECHA about the eligibility for a fee reduction in accordance with Article 13(1) of Regulation (EC) No 340/2008. Ceasing to manufacture or import of substances have no consequence on that obligation.
If after registering a substance, you have decided to stop manufacturing or importing it, you should still pay the applicable registration fee in full in case ECHA found that you were not eligible for the claimed SME fee reductions. Once the registration fee is paid and, therefore, your registration can be considered complete, you can indicate in REACH-IT that you have ceased the production or import of a substance. If at a later stage you decide to restart manufacturing or importing it, you would only need to report it again in REACH-IT and the registration would still be valid. Otherwise, you would need to submit a new registration and pay a new registration fee.
In case the balance to the correct registration fee is not paid, such registration will be de-activated in ECHA’s database and the rejection of the registration will be communicated to you by a decision.
The SME verification process relates to your information obligations at the time of submitting your registration, which includes the obligation to inform ECHA about the eligibility for a fee reduction in accordance with Article 13(1) of Regulation (EC) No 340/2008. The termination of the OR agreement has no consequence on that obligation.
Since your represented company benefited from reduced SME fee at the time of registration, therefore you, as the appointed OR at that time, are still liable to demonstrate the eligibility for the SME fee reduction obtained, once ECHA initiates the verification of your represented company size. The failure to provide complete documentation would result in ECHA finding your represented enterprise as non-eligible for the claimed SME fee reductions. In such case, the OR would be liable for the difference between the fee already paid and the correct registration fee, as well as the applicable administrative charge.
Yes. A notification under Directive 67/548/EEC is nominal so that only the notifier benefits from it under REACH. Therefore, any other parties manufacturing or importing the substance in quantities of more than one tonne per year, must register it, unless another registration exemption applies.
More information on notified substances can be found in the Guidance on registration, article 24 (2) of REACH and in section 2.2.4.3- Notified substances according to Directive 67/548/EEC.
Yes. Unclaimed notifications are considered registered under REACH and therefore the notifiers are listed as registrants in the co-registrant’s page.
In accordance with Article 24 of REACH, ECHA assigned a registration number to each notification submitted under Directive 67/548/EEC and the substance is considered as registered. As for any other substance registered under REACH, both the name and the country of the registrant will be available in the co-registrant’s page. If a potential registrant submits an inquiry for this substance to ECHA, they will find this information, according to Article 26(3) of REACH.
As a transitional measure after the entry into force of the REACH Regulation in 2007, ECHA set up a process allowing legal entities who had notified a substance under Directive 67/548/EEC to communicate to the Agency their administrative details and claim the REACH registration number assigned to their notification.
In light of the time passed since then, the possibility to claim the registration numbers assigned to NONS notifications has been discontinued. This does not affect the registration numbers already claimed before the process ended.
From now on, if you intend to manufacture or import a substance previously notified under Directive 67/548/EEC in quantities of 1 tonne per year or above but have not claimed a REACH registration number, you should follow the registration process set out under REACH.
For a handful of NONS notifications, the tonnage band may be incorrect in REACH-IT and the system may be unable to accept an update of the registration dossier or might issue a wrong invoice.
Under REACH there are three possible registration types: standard registration, transported isolated intermediate, and on-site isolated intermediate. For each one of them there is a different registration fee.
Under Directive 67/548/EEC there was no such specific distinction and only one type of standard notification was required. All NONS notifications are initially regarded as standard registrations in REACH-IT and have the same tonnage band for standard and transported isolated intermediate.
It is extremely important and very easy to check that the tonnage band of your notification is correctly indicated in REACH-IT after claiming your registration number and before you submit any update of your registration dossier. You should make sure that the maximum tonnage band displayed under ‘Dossier type’ and ‘Payment information’ matches the tonnage of your NONS notification.
Information on the tonnage band of a registration is displayed in the reference number page.
If you see any mistake in the tonnage band shown in reference number page, you should follow the instructions of Q&A 0689.
You need to contact your relevant Member State Competent Authorities. They will in turn confirm that an amendment is necessary and provide ECHA with the correct information.
You can find the contact details of all the Member State Competent Authorities here.
Detailed information on the update process for NONS registrations can be found at How to update your previously notified substance (NONS), point 4. Migrate your scientific information to IUCLID 6.
The registration dossier must be updated if one or more of the update reasons for registrations under REACH applies (Article 22 and Article 24(2)). For example, a change in the composition of the substance, the status of the registrant, the classification and labelling, or if the annual quantities manufactured or imported reach the next tonnage band.
It is important to verify the information available for your notification in REACH-IT before you start the dossier update. For more details, see How to prepare registration and PPORD dossiers, Annex 4. Minimum information required for updating a registration under previous Directive 67.
Find more information on dossier updates at Keeping your dossier up to date.
Your dossier will undergo a series of initial administrative checks called "business rules". ECHA accepts a dossier for processing only if all the relevant business rules are satisfied.
Once your dossier has been submitted:
- It will be allocated a submission number.
- It will undergo a completeness check.
- You will receive an invoice if there the tonnage band has been upgraded or if you have added chargeable confidentiality claims not made under Directive 67/548/EEC.
It will be regarded as complete once ECHA has verified the completeness of the information you submitted and received the payment of the relevant fee (Article 20(2) of REACH).
In the case of an update of a registration to comply with an evaluation decision issued in accordance with Article 135 of REACH, the submitted information will be evaluated by ECHA pursuant to Article 42 or by the requesting MSCA pursuant to Article 48, depending on the legal basis of the original request of the MSCA under Directive 67/548/EEC.
Yes, substances notified under Directive 67/548/EEC (NONS) need to be classified, labelled and packaged in accordance with the CLP Regulation. CLP repealed this Directive with effect 1 June 2015 (Article 60).
These substances are regarded as registered under REACH. Therefore, their classification and labelling information must be included in the C&L Inventory. After a registration number has been claimed by the NONS notifier, the respective registration dossier must be updated with the CLP classifications without undue delay, and a separate notification to the Inventory is not required.
For NONS notified below one tonne under Directive 67/548/EEC and for which no tonnage band update has been done, a separate notification to the Inventory will have to be submitted if the substance is classified as hazardous and is placed on the market. So, if the annual volume of the NONS substance remains below one tonne, the company must submit a C&L notification. When the annual volume has reached or exceeds the one tonne threshold, an update in the form of a registration dossier is required.
Yes. REACH requires that certain information on chemical substances is made available to the public. This concerns the information specified in Article 119 of REACH. You can search for publicly available information on NONS at the information on chemicals database.
On the ECHA website NONS dossiers can be recognised by their purple background while other registration dossiers have a blue background.
For further information, see Dissemination and Confidentiality under the REACH Regulation, Chapter 2.5, Publication and confidentiality of NONS, page 16.
Under the NONS Directive you could claim confidentiality on:
- Tonnage band;
- (Robust) Study summary; and
- IUPAC name of the substance.
Confidentiality claims accepted under Directive 67/548/EEC remain valid under REACH, in as far as the information has in the meantime not become available in the public domain. ECHA will assess the confidentiality claim and will – if the information is found to be publicly available – request further information before rejecting the claim.
You do not need to pay for claims that were already made under Directive 67/548/EEC. For more information, see How to update your previously notified substance (NONS)
For the case where confidentiality of the IUPAC name was requested under Directive 67/548, but the IUPAC information in the meantime is already available in the published EC Inventory or at any other publicly available source, ECHA assumes the request to be expired, unless the registrant provides a full justification that includes a valid reason why the information should still be kept confidential despite of the public availability.
Under REACH, it is foreseen that also the following information is published on the ECHA website, unless claimed confidential:
- Information contained in the safety data sheet (including name of the notifier, registration number, uses and uses advised against);
- Trade name of the substance;
- If essential to classification and labelling, the degree of purity of the substance and the identity of impurities and/or additives which are known to be dangerous.
Further information is available in the manual on Dissemination and Confidentiality under the REACH Regulation.
ECHA gives the same possibilities to NONS notifiers as to other REACH registrants for claiming confidentiality. Therefore, if your company considers that this information should not be published on the ECHA website, you can introduce the relevant confidentiality claims. These confidentiality claims need to be justified and ECHA will charge a fee for each claim, in accordance with Article 119(2).
ECHA will assess the confidentiality claim and will – if the information is found to be publicly available or if the claim is not sufficiently justified – request further information before rejecting the claim.
Further information can be found in the NONS Question and Answers.
If a company purchases from an EU supplier a substance in a non-nanoform and mills it down to generate a nanoform of the substance, this particular activity is considered as a downstream use of the substance, rather than as manufacturing of the substance. Therefore, as a downstream user you do not have registration obligations, but you are subject to the downstream users’ obligations set out in Articles 37 and 38. For further information please see the Q&As on Nanoforms of substances, chapter I. Downstream user obligations.
The REACH Regulation states that the tonnage band is calculated per registrant per substance, including non-nanoforms and nanoforms. Thus, the registration obligation depends on the total yearly tonnage of the substance that you manufacture or import, including both non-nanoforms and nanoforms. If the overall volume of the substance is above 1 tonne per year, nanoforms within that total volume also need to be registered, even if they represent a volume of less than 1 tonne per year.
Similarly, the overall quantity of the substance, cumulating the quantity of both non-nanoforms and nanoforms, determines the applicable information requirements. The information requirements of the total volume apply to the non-nanoform and each of the nanoforms of the substance, even if their individual volumes represent a lower tonnage band.
For more information see the Appendix for nanoforms applicable to the Guidance on Registration and Substance Identification, section 2.1 Registration obligations.
Yes, a polymer can also meet the definition of a nanoform.
The obligation to submit information on nanoforms as set out in REACH Annexes VI to X only applies to substances subject to registration. Polymers are exempt from registration. Nevertheless, any manufacturer or importer of a polymer shall submit a registration for the monomer substance(s) or any other substance(s) that have not already been registered by an actor up the supply chain, if the conditions set out in Article 6(3) of REACH are met.
For specific cases we recommend contacting our helpdesk for a more detailed reply.
Before being introduced into the polymer during the polymerisation phase, the nanoform has to be manufactured or imported. If you manufacture or import in a quantity above 1 tonne per year the substance of which nanoform is introduced in the polymer, you must register the nanoform, even if it is then used in the manufacturing of a polymer.
However, if you buy a nanoform from a supplier in the EU, you do not have to register it, but the manufacturer or importer in the supply chain must do so. In the latter case, you have to ensure that your use of the nanoform is covered by the registration of the manufacturer/importer.
Finally, if you import the polymer you must submit a registration for the nanoform if it has not already been registered by an actor up the supply chain and if the conditions set out in Article 6(3) of REACH are met (see Q&A 1674).
Each registrant is responsible for registering the nanoforms that they themselves manufacture or import. The information required by Annex VI, including the characterisation of nanoforms or sets of nanoforms, must be submitted separately by each registrant.
The information required by Annexes VII-X can be submitted jointly in the lead registrant dossier on behalf of the member registrants. Alternatively, this information can be submitted separately by each registrant via the opt-out mechanism.
If a co-registrant claims not to produce nanoforms but does not provide any proof to support their claim, the other registrants of the substance should proceed with the registration of their nanoforms following the REACH requirements.
Registrants that manufacture/import nanoforms of the substance after 1 January 2020 without a registration that covers these nanoforms are in breach with their duties under the REACH regulation, which may lead to enforcement measures by national enforcement authorities.
If none of the characterisation parameters of the nanoform(s)/set(s) of nanoforms reported in the existing registration dossier change, there is no need to update the dossier with regard to the identity of the nanoforms covered.
Note that even if none of the numerical characterisation parameters change, if the change in the production line concerns the surface-treatment process, the description of the surface-treatment process in the registration dossier may still need to be updated.
A batch-to-batch variability results from the variation of process parameters inherent to a manufacturing process that is defined by a series of parameters (e.g. starting materials, solvents, temperature, order of manufacturing steps, etc.).
If the variation in a parameter is only due to batch-to-batch variability, then these are still considered as being the same nanoform. However, if there are deliberate changes to the process parameters, then these are different nanoforms.
- The registrant reports clearly defined boundaries in terms of characterisation parameters of the nanoforms, which are part of the set
- The registrant justifies that the hazard, exposure and risk assessment of the nanoforms can be performed jointly. This justification is substantiated by supporting data on the nanoforms in the set.
The REACH Regulation defines a substance as “a chemical element and its compounds”. A substance becomes a different substance when there is a modification of the chemical element or the compounds that compose it. Following from this, the modification of the form of a substance, including its modification into a nanoform, does not make it a different substance under REACH.
REACH also specifies that the surface treatment of a nanoform does not lead to the creation of a new substance, but a new nanoform of the substance concerned. Each nanoform must be specifically characterised and reported in the registration dossier.
There is no definition for the term “granular form" in REACH. However, this can be interpreted to mean "particles".
All nanoforms, by definition, will have at least 50% of their particles by number below 100 nm, and all nanoforms will be "granular" when available as a dry powder. However, some nanoforms are available only in suspensions, or are incorporated into a matrix throughout their entire lifecycle. In this case, there may be no exposure to the dry powder, or the granular form in general.
- The justification must address separately each characteriser listed in section 2.4 of Annex VI. To this end, it must follow the structure of the text template available in the IUCLID section 1.2 field 'Justification for reporting set of similar nanoforms' and address all applicable points in it.
- The justification must be substantiated by scientific evidence addressing the physicochemical, environmental fate, ecotoxicity and toxicity properties of nanoforms that are within the boundaries of the set of nanoforms. For each characteriser, the justification must summarise the supporting data.
- Each scientific evidence summarised in the justification must be submitted in the form of a (robust) study summary
In all the above cases, the findings from these studies and the characterisers of the nanoforms used in the studies must be summarised in accordance with the points listed.- The (robust) study summary can be provided by attaching it in the section 1.2 field ‘Attached information’;
- When the (robust) study summary is reported elsewhere in the IUCLID dossier, e.g. in IUCLID section 4-7, a reference to it can also be made by linking it via the section 1.2 field ‘Cross-reference’;
- Alternatively, references to publicly available literature may be provided instead of a robust study summary.
- For each characteriser, the justification must explain how the scientific evidence demonstrates that all the nanoforms in the set can be assessed jointly. This explanation must clearly show that the nanoforms used to generate the supporting data are representative of all the nanoforms included in the boundaries of the set.
- The justification must always be provided in full in the field 'Justification for reporting set of similar nanoforms' or in the field ‘Attached information’. The justification cannot refer to explanations provided elsewhere in the dossier.
If the registration will be updated to cover additional nanoforms, the decision needs to be made whether the nanoforms will be registered (i) on their own as separate nanoforms; (ii) as a new set of nanoforms; or (iii) by modifying the already registered set of nanoforms.
If the nanoforms are added to the registration as separate nanoforms or as a new set of nanoforms, they will not impact the already registered set of nanoforms. Similarly as for the existing set, they need to be registered by including in the dossier the appropriate characterisation of the set, the set justification and the Annex VII-X information corresponding to the set.
If the nanoforms are added to the registration in an existing set of nanoforms, you need to ensure that the nanoforms fit within the clearly defined boundaries of characterisers of the existing set. If this is not the case, you need to analyse whether the boundaries of the set can be expanded without affecting the joint hazard assessment, exposure assessment and risk assessment of all the nanoforms covered by the set. This analysis must be reflected in the provided justification for the set.
The principles described in the Appendix for nanoforms applicable to the Guidance on Registration and Substance Identification for forming a set of nanoforms give advice to the registrants on how to build a set of nanoforms. Following the principles in the Guidance reduces the need for development of complex justifications for the set.
The limitations for the formation of sets of nanoforms are based on presumptions resulting from a scientific consensus. The stronger the consensus, the stricter the limitation. For the strictest restrictions (e.g. fibres and sphere shapes must be addressed in different sets of nanoforms), the scientific consensus is such that the presumption is considered not rebuttable.
For other restrictions, deviating from the principles set in the Guidance will increase the complexity of a justification as well as the supporting evidence required, and this will increase the level of scrutiny given to the justification.
In any case, for each set of nanoforms a robust justification that the hazard, exposure and risk assessment can be performed jointly for all nanoforms in the set must always be provided. It is the responsibility of a registrant to develop the justification and provide the data supporting the justification.
The completeness of the justification is checked during the technical completeness check and its compliance in a potential compliance check.
In order to create a set of nanoforms, you need to clearly define the boundaries of your set of nanoforms, and provide a justification to demonstrate that the hazard assessment, exposure assessment and risk assessment of the nanoforms in the set can be performed jointly for all nanoforms included in the set, without exceptions. This demonstration may require you to test some of these nanoforms. Indeed, this justification must be substantiated by scientific evidence addressing the physicochemical, environmental fate, ecotoxicity and toxicity properties of nanoforms that are within the boundaries of the set of nanoforms. Each scientific evidence summarised in the justification should correspond to a (robust) study summary. For further information on how to justify a set of nanoforms, refer to Q&A 1681.
Once a set of nanoforms has been established and scientifically justified, the complete Annex VII-X information must be provided for the set of nanoforms. It is not necessary to generate data on the Annex VII-X properties for each nanoform included in the set. Indeed, as mentioned above, the justification for the set of nanoform must prove that the hazard, exposure and risk assessments of the nanoforms can be performed jointly for all nanoforms in the set. However, the data provided for each information requirement must be representative for the whole set of nanoforms.
For every information requirement as per Annex VII-X, the registrant must submit either (i) studies performed on the nanoforms concerned; or (ii) studies on other forms of the substance accompanied by endpoint-specific justifications as to why this information is adequate for assessing the nanoforms concerned; or (iii) relevant adaptations as foreseen by Annex XI of REACH or Column 2 of the relevant Annex VII-X.
As explained in chapter G. Joint submission, the Annex VII-X information can be submitted by the lead registrant on behalf of the other assenting registrants, or separately by co-registrants via the opt-out mechanism.
The legal requirement under Annex VI of REACH is to report the number-based particle size distribution with indication of the number fraction of constituent particles in the size range within 1-100 nm. You are free to choose the measurement method as long as the data requirement is fulfilled. We recommend using at least one electron microscopy technique.
If laser diffraction can provide that information, it can be used. In many cases, this may not be the case. All the measured particles must be taken into consideration when the number fraction of constituent particles is determined.
The requirement in REACH Annex VI is to report the number-based particle size distribution with indication of the number fraction of constituent particles in the size range within 1-100 nm. This is the case even if the constituent particles of the nanoform you manufacture/import form agglomerates. This information is reported by each registrant in sections 1.2 and 1.4 of IUCLID .
Information on the size of the agglomerates shall be reported in IUCLID section 4.5 “Particle size distribution (Granulometry)”. The information in section 4.5 is normally submitted jointly by the lead registrant on behalf of the co-registrants. Nevertheless, it can be submitted by a member registrant via the opt-out approach, especially if the information submitted is specific to him and considered confidential.
- The registrant reports clearly defined boundaries in terms of characterisation parameters of the nanoforms, which are part of the set;
- The registrant justifies that the hazard, exposure and risk assessment of the nanoforms can be performed jointly. The justification must be supported by data on the nanoforms in the set. For more information on the set justification, refer to Q&A 1681.
The completeness check ensures that all the required information is reported in the dossier. Therefore, you cannot pass the completeness check by providing placeholders for missing information, even if the tests have been commissioned. If you cannot address the information requirement with available data or an adaptation (or, in the case of Annex IX-X information requirements, with a testing proposal), you will not pass the completeness check for this information requirement.
If you fail the completeness check, you will be given a standard deadline of four months to provide the missing/incomplete information. Therefore, if you fail the completeness check at first attempt and in the meantime obtain the test results, you can amend the dossier and pass the completeness check at second attempt. The submission date will correspond to the submission of your first attempt.
For advice on how to report unfinished tests that were commissioned in response to an ECHA (draft) requesting you to carry out a test, please refer to the document Information on manual verification at completeness check, chapter 2.
You can only submit one registration dossier per substance. Therefore, all the forms, including nanoforms, of the same substance must be covered by one single registration dossier. This means that you will have a single registration number for the registration of the substance, covering both the non-nanoform and nanoform(s).
A surface-treated and a non-surface treated nanoform are two separate nanoforms that you will need to characterise and report separately in your registration dossier, with a corresponding hazard dataset for each nanoform.
If you use the approach to register individual nanoforms via a set of nanoforms, it is important to note that where both surface-treated and non-surface-treated nanoforms are covered by a registration, surface treated and non-surface-treated nanoforms must a priori not be included in the same set of nanoforms.
To be able to build the set(s) of nanoforms, you may need to characterise the individual nanoforms in order to allocate them in the appropriate set and provide an adequate justification.
If nanoforms are included in a set of nanoforms, you are not expected to report information on the characterisation of all the nanoforms in the set in the IUCLID dossier. Instead, when registering nanoforms via a set of similar nanoforms, you must clearly report the boundaries of the set for each of the characterisers and provide a justification for the set, per characteriser.
To register a nanoform of a substance, you must report its characterisation parameters in sections 1.2 and 1.4 of IUCLID, as defined by REACH Annex VI. The information reported in these sections refers to the identification of the compositions/forms of the substance within the scope of the registration. It is submitted separately by each registrant.
The Annex VII-X information requirements for the nanoform are then reported in sections 4-7 of IUCLID. This information can be submitted jointly by the lead registrant on behalf of the co-registrants; or separately by co-registrants via the opt-out mechanism.
While IUCLID section 4.28 contains the templates for reporting (robust) study summaries of certain physicochemical properties for nanomaterials, the only sub-section in 4.28 that corresponds to a mandatory Annex VII-X information requirement is section 4.28.8 – Nanomaterial dustiness. The remaining sub-sections under 4.28 do not need to be provided as part of a registration dossier.
- If the information is jointly submitted, the co-registrant only has to identify his nanoform(s) under Annex VI, but he does not have to submit himself the hazard data that cover his nanoform(s). Instead, the co-registrant must link in his registration dossier his nanoform(s) to the corresponding hazard data submitted in the lead dossier. To this end, the co-registrant must refer to a boundary composition record of the lead dossier in the field ‘Reference to related composition(s)’ of each section 1.2 legal entity composition record. This boundary composition must be relevant to the co-registrant’s nanoform or set of nanoforms, and must be linked to the Annex VII-X information submitted to cover the information requirements for it.
- Note that if the jointly submitted information concerns a set of nanoforms, the co-registrant must ensure that his own nanoform(s) fall within the characterisers reported for a boundary composition for a set of nanoforms in the lead dossier, and that the justification provided for the set of nanoforms in this same boundary composition covers his nanoforms.
- If the information is submitted separately by the co-registrant, he must ensure that a specific hazard data set is provided for his nanoform(s) via an opt-out, as foreseen in Article 11(3) of REACH. In this second case, the co-registrant must submit all the hazard data required for his nanoforms in sections 4-7 of IUCLID.
If you have submitted your member dossier but rely on the lead dossier for Annex VII-X information for your nanoform of the substance, ECHA could not verify the completeness of this data. Therefore, ECHA reserves the right to assess the completeness of your dossier in relation to the information on the Annex VII-X requirements, whenever this information is submitted by the lead registrant.
Once the lead registrant has submitted their dossier, you must update your registration to link your nanoforms with the relevant Annex VII-X information in the lead dossier. This is done by referring to the corresponding boundary composition name(s) of the lead dossier, in the section 1.2 Legal entity compositions of your own dossier by using the field ‘Reference to related composition(s)’.
Yes. Due to the differences between non-nanoforms and nanoforms you need to demonstrate for each endpoint and each nanoform why the information generated on the non-nanoform of the substance is adequate for assessing the nanoform, i.e. that the prediction from the non-nanoform to each nanoform is reliable and accurate, and does not underestimate the hazard.
To report the read-across in your dossier, follow the advice given in the manual “How to prepare registration and PPORD dossiers”, chapter 9.6.3 ‘How to report read-across in IUCLID'.
Downstream user obligations under Article 37(4) are triggered upon receipt of a safety data sheet (SDS) with a registration number (Article 39(1) of REACH). As a downstream user, you receive an SDS with exposure scenarios attached for a hazardous substance (including nanoforms) registered above 10 tonnes/year.
When receiving an SDS, as a downstream user you must establish whether your use and/or your conditions of use are covered by the exposure scenario information in the SDS and take action if that is not the case.
When you are dealing with a nanoform (that you have purchased or created yourself), ensuring that the use is adequately covered requires checking both the uses and conditions of use in the exposure scenario(s), and whether the nanoform you handle is covered by the extended SDS.
When downstream users apply a surface treatment to a nanoform they purchased or create a nanoform by milling a non-nanoform (see FAQ 1838), they must check whether:
• the nanoform created is covered by the SDS received (see Q&A 1831 and Q&A 1833) and
• the use (e.g. the process of surface treatment or the milling) is covered by the exposure scenario(s) (see Q&A 1834).
Q&A 1834 explains the actions to be taken when you conclude that your uses or the nanoform you create is not covered.
If your uses and any nanoform you create are covered in the exposure scenario, no further action under REACH is needed. Document your check and the actions you have taken to reach your conclusion and make this information available to enforcement authorities upon request. Clear documentation helps you to justify your assumptions in a transparent way and helps the authorities to better understand the criteria you have applied in your decisions.
The general duties of REACH applicable to downstream users are described in the Guidance for downstream users.
A nanoform must be characterised in accordance with section 2.4 of Annex VI to REACH. A substance may have one or more different nanoforms, based on differences in the size distribution, shape and other morphological characterisation, surface treatment and functionalisation and specific surface area (SSA) of the particles. Variation in one or several of these characterisers results in a new nanoform.
In practice, if you modify any of these characterisers while using the nanoform that you have purchased (e.g. by grinding, applying a surface treatment) or if you create a nanoform from a non-nanoform (e.g. by milling the non-nanoform, see QA 1838) you must check whether your supplier has covered the resulting nanoform in their extended safety data sheet (SDS) (assuming one has been received). To perform such a check, you must have enough information on the characterisers of the nanoforms you have created (See Q&A 1832).
If the nanoform you create is covered in the exposure scenario, no further action needs to be taken in this respect (but you must still check if your uses of the nanoform supplied are covered, see Q&A 1833). You must document your check and the actions you have taken to reach your conclusion and make this information available to enforcement authorities upon request. Clear documentation helps you to justify your assumptions in a transparent way and helps the authority to better understand the criteria you have applied in your decisions.
As a downstream user, you receive an SDS with exposure scenarios attached for a hazardous substance (including nanoforms) registered above 10 tonnes/year. If your supplier is covering the uses of your nanoform (including where relevant the creation of a nanoform and its subsequent uses), then the SDS will indicate these uses in Section 1.2 and will include the associated exposure scenario(s) annexed that describe the conditions of that use. The supplier’s SDS must also include the nanoform characterisation information.
REACH Annex II, which prescribes the contents of an SDS has recently been updated. Until 31 December 2022, an SDS may continue to be provided in accordance with Regulation (EU) 2015/830 or can be provided in the new format, according to Regulation (EU) 2020/878. If the supplier’s SDS has been compiled in accordance with Regulation (EU) 2020/878, then the nanoform characterisation information will be found in Section 3 (3.1 for substances, 3.2 for mixtures) or Section 9. If the SDS has been compiled in accordance with Regulation (EU) 2015/830, the supplier can include the information in Section 9 under the subsection ‘(a) Appearance’, where information on “granulometry” should be included. For further details on obligations of SDS suppliers, please see the Guidance on the compilation of safety data sheets.
If you create a nanoform as a downstream user, and the SDS you have received from your supplier contains characterisation information in terms of size, shape, surface treatment and specific surface area (SSA), you must compare it with the information on the nanoform that you have generated and decide whether or not your nanoform is covered by the supplier’s SDS. The Guidance ‘Appendix for nanoforms to the Guidance on Registration and Substance Identification’ provides details on how you can distinguish between different nanoforms. In practice, the information you receive in the SDS may refer to a single nanoform with characterisation data for each of the four characterisers (size, shape, surface treatment and SSA) (see example of possible format in Table 1 on p. 50 of the Guidance on the compilation of safety data sheets). In that case, you can consult section 4 of the ECHA Guidance ‘Appendix for nanoforms to the Guidance on Registration and Substance Identification’, in particular the subsection “Principles on the boundaries of sets of nanoforms” dedicated to each of the nanoform characterisation parameters to make a decision on whether your nanoform is covered by applying the same principles outlined in the guidance.
In the case that the supplier’s SDS refers to a set of nanoforms, the boundaries covered for each of the parameters should be specified in their SDS and allow you to decide whether the nanoform you have created is covered. In such a case, all the characterisation parameters of your nanoform must fall within the boundaries set in the SDS to ensure that it is covered.
Downstream users’ obligations are triggered upon receipt of an (extended) SDS with a registration number. There may be genuine reasons where an SDS is not required to be supplied by law, e.g. because the substance (and nanoforms) are not classified under CLP, the substance is not classified as PBT/vPvB, and/or the substance is not on the REACH Candidate List. There are also genuine reasons where there may be no exposure scenarios annexed to the SDS, e.g. because the registration tonnage is below 10 tonnes/year, or the substance is registered for use only as an intermediate under strictly controlled conditions. If you have been supplied a substance (including as a nanoform) without an SDS, or with an SDS but without an exposure scenario and you think you should have received one, it is recommended that you formally communicate with your supplier in writing to check the reason for not having received an SDS and document this action.
Where your supplier is not required to provide an SDS, you may still have some obligations under REACH as a downstream user if you modify the nanoform you have purchased, or you create a nanoform from a non-nanoform of the substance. If, for example, the nanoform you create fulfils the criteria to be classified as hazardous (you may need testing to determine whether classification applies), you must generate an SDS for the safe use of that nanoform at company level (for your own workers) if you meet the conditions set out in Article 35 of REACH (or grant access to your workers to the same information as provided in an SDS). If you put the nanoform created on the market, becoming a supplier, you also have to provide an SDS to your customers according to Article 31 of REACH. You also need to inform upstream to your supplier, according to Article 34, that the nanoform is hazardous.
Where your supplier has provided an SDS but no exposure scenario covers your use(s), REACH gives a downstream user the right to make a use (including the creation or use of a nanoform) known to their supplier (Article 37(2) of REACH). You must provide sufficient information to your supplier to enable them to assess your use, as described in Q&A 1835. When the supplier includes that use in their REACH registration, the supplier must then create/update the SDS they provide to their downstream user customers, to enable the customers to comply with REACH Articles 35 and 31.
The process of checking the uses and conditions of use is not different for nanoforms and non-nanoforms. Be careful that you check the uses applicable to your nanoform (in case the safety data sheet (SDS) covers several (nano)forms of the substance). Details on how to perform the check (on whether your uses and conditions of use are covered by the provided exposure scenario) are provided in section 4.2 of the Guidance for downstream users. The check includes three steps:
- Checking the use(s): compare your use(s) with the “identified use(s)” in SDS Section 1.2 and the title section of the attached exposure scenarios;
- Checking processes/activities of the exposure scenario: compare your processes/activities with those described in the exposure scenario to ensure that the way you use the substance has been covered in the supplier’s assessment;
- Checking the conditions of use: compare your onsite operational conditions and risk management measures (including their effectiveness) with those described in the exposure scenario(s).
If you conclude that your use is covered by the exposure scenario received, no further action under REACH is needed. You should nevertheless document your check and any action you take to guarantee your compliance with the conditions of use in the exposure scenario.
If you conclude that the nanoform you have created or its uses are not covered by the information in your supplier’s safety data sheet (SDS), there are three options available to you:
- Stop the uses and the creation of the nanoform that is not covered.
- Ask your supplier to include the nanoform you have created, as well as the use(s) or conditions of use in their chemical safety report and to provide you with an exposure scenario for it (see Q&A 1834). In this case, you must make sufficient information available to your supplier to enable them to make an assessment. In the case that you are requesting a new nanoform to be covered, this information should normally include the characterisation of the nanoform (Annex VI characterisers) unless your supplier agrees to generate this information for you. If new hazard data needs to be generated, you may agree with your supplier on who is performing the testing and/or covering the costs.
If your supplier refuses to cover your uses/ your nanoform or if the supplier advices against your use/ your nanoform, you can try to find another supplier that covers your uses/ nanoform. When looking for a possible supplier that can cover your nanoform, it may be useful to consult the ECHA dissemination web pages to check whether your nanoform is covered by the registration dossier of the substance to get an idea on how likely it is to find a supplier that covers your nanoform. - Alternatively, you can cover the nanoform or uses yourself. In this case, you must carry out your own chemical safety assessment and prepare your own downstream user chemical safety report (DU CSR) for your uses and conditions of use, unless exemptions apply (Article 37, (c) use <1 tonne/year or (f) PPORD use). When you perform a DU CSR, or you are relying on exemptions, you must notify this to ECHA (see Q&A 1837).
More information on how to handle exposure scenarios can be found in: Practical guide 13: “How downstream users can handle exposure scenarios”.
Practical Guide 17: “How to prepare a downstream user chemical safety report” contains practical information on how to perform a downstream user chemical safety report (DU CSR). When you perform a DU CSR for a nanoform, it is advisable to follow the steps detailed in the guide, taking into account the specificities for nanoforms detailed below to determine which of the three approaches explained you should follow.
The content of the DU CSR would depend on whether you need to assess and describe:
- a new nanoform (created from bulk or modified from a nanoform), or
- only new uses or new conditions of use of a nanoform already covered by an exposure scenario, or
- both a new nanoform and new uses and conditions of use.
Whenever there is a new nanoform, it is likely that you will have to follow Approach C in the Practical guide (which includes reviewing the hazard assessment).
Information on the nanoforms
If you conclude that the nanoform you created is covered by the information in your supplier’s safety data sheet (SDS), you can follow the advice of the section below (information on uses and conditions of use).
If you conclude that the nanoform you have created is not covered and you intend to assess it by yourself, you must determine whether the hazard assessment performed by your supplier is applicable to your nanoform.
You cannot simply presume that your nanoform is covered by the hazard assessment performed by the supplier. You must have a justification on why this is the case, document your conclusion and make it available to enforcement authorities upon request (see principles below).
The Guidance ‘Appendix for nanoforms to the Guidance on Registration and Substance Identification’, provides advice in section 4 on a set of principles that helps to define the boundaries of a set of nanoforms, where variation (in the characterisation parameters in 2.4.2 to 2.4.5 of REACH Annex VI) within these boundaries does not affect the hazard assessment. The same principles could be applied by a downstream user (you) to determine whether the nanoform you have created and its hazard assessment falls within the boundaries of your supplier’s nanoform/ hazard assessment. The Guidance also gives some indication of what must be considered to justify that the hazard assessment performed by the supplier covers your nanoform.
If, after following the above principles, you cannot conclude that the hazard assessment from your supplier is applicable to your nanoform, you must perform a hazard assessment for your nanoform. Though not an explicit requirement, the information requirements as specified under Annexes VII to X would be expected to be followed in order to perform the hazard assessment on the nanoform.
You have the possibility to apply read-across or other adaptations to address hazard endpoints related to your nanoform. The ‘Appendix R.6-1 for nanoforms applicable to the Guidance on QSARs and Grouping of Chemicals’ gives advice on how to justify the use of hazard data between nanoforms and/or sets of nanoforms, and the non-nanoforms (bulk) of the same substance.
If the information required to conclude on the hazard assessment can only be obtained by testing, you should follow the advice provided in the three nano-specific appendices to the Guidance on IR&CSA (endpoint specific Guidance), Chapter R.7a, R.7b and R.7c. Where this information can only be obtained by testing for Annex IX and X standard information requirements you must submit a proposal for a testing strategy to the Agency in accordance with Article 38.
Information on use and conditions of use
If, when checking the uses identified by your supplier or the conditions of use applicable to the uses, you conclude that (some) are not covered (see Q&A 1833) you must perform this step. This involves the identification of uses and conditions of use that are not covered by the information in your supplier’s SDS and the generation of exposure scenario for these uses.
The advice in the Practical Guide 17: “How to prepare a downstream user chemical safety report” can be followed. However, attention should be paid to the following:
- When the uses have been identified and assessed by the supplier but you have determined that your nanoform is different, you cannot assume that the exposure potential/ (e.g. dustiness) of the nanoform you have created is the same. The exposure estimation may need refinement.
- When performing the exposure estimation step, it should be noted that the method adopted (either direct measurement or an exposure estimation modelling tool) must be applicable to the nanoforms.
If you need to prepare a downstream user chemical safety report (DU CSR) or you rely on the exemptions in Article 37(4)(c) or (f), then you must report certain information to ECHA.
A downstream user of nanoforms can report information to ECHA via IUCLID, as described in the manual on How to prepare a downstream user report.
The reporting comprises the following required information (the corresponding IUCLID section is indicated in brackets):
- Identity of the downstream user, i.e. name, contact details (REACH-IT account and IUCLID section 1.1 Identification);
- Identity of the substance and characterisation of the nanoform as specified in section 2.4 of Annex VI to REACH (IUCLID sections 1.1 Identification and 1.2 Composition)
- The registration number(s) as communicated to the downstream user by their suppliers (IUCLID section 1.3 Identifiers);
- Identity of the manufacturer(s), importer(s) or other supplier, i.e. name, contact details (IUCLID section 1.7 Suppliers);
- Site(s) of use (IUCLID section 3.3 Sites);
- A brief general description of the use(s) (IUCLID section 3.5 Life Cycle description) including the information on the conditions of use(s);
- If appropriate according to Article 38(2)(f), a proposal for additional testing on vertebrate animals (relevant endpoint study record(s) in IUCLID section 5-7);
- An indication of the type of report (including any exemptions relied upon), and an explanation for why the report was required, including why the issue could not be resolved through the supplier (IUCLID Section 14).
A non-EEA company (that can appoint an only representative, see FAQ ID=15) may, by mutual agreement, appoint a natural or legal person established in the European Economic Area (EEA) to act as his only representative. According to Article 8(2) of REACH this representative shall comply with all obligations of importers under REACH. Therefore the only representative is required to have sufficient background in the practical handling of substances and the information related to them. More information on the only representative is also provided in section 2.1.2.5- 'Only representative of a "non-EU manufacturer"' of the Guidance on registration: http://echa.europa.eu/guidance-documents/guidance-on-reach.
Yes, an only representative can represent one or several non-EU companies that manufacture substances, formulate mixtures or produce articles which are exported to the European Economic Area (EEA), even for the same substance.
If an OR is appointed by several non-EU companies, then the OR needs to create several OR accounts in REACH-IT, one for each non-EU company they represent. There should be a 1-to-1 correspondence between an OR account in REACH-IT and the non-EU company they represent. If the same legal entity covers the role of a manufacturer/importer and the role of an OR, then they will need to create two different accounts in REACH-IT: one account for their role as manufacturer/importer and another account for their role as OR.
More information on the duties of the only representative is provided in section 2.1.2.5- 'Only representative of a "non-EU manufacturer"' of the Guidance on registration.EU importers of a non-EU manufacturer can apply for an authorisation irrespective of whether they are covered by an OR for registration of the Annex XIV substance. If the appointment of the OR does not extend to also cover the fulfilment of the obligations of the importers with regard to authorisation, then the importers themselves can apply for an authorisation. In such a case, the importers will apply in their role as importers of an Annex XIV substance. Furthermore, the importers are not required to indicate the name of the non-EU manufacturer or the OR in their application for authorisation sent to ECHA, since the OR's appointment and their respective obligations are limited to the registration of the Annex XIV substance.
Yes, you need to notify ECHA about this change. You represent the non-EU manufacturer and, therefore, you need to communicate changes in their legal personality. You can notify ECHA using the legal entity change functionality in REACH-IT.
To change the legal personality in this case you need to:
- Create a new account in REACH-IT for your company to represent the new legal entity, reflecting their size. Note that in the "Company Size" information, you must indicate the size of the non-EU/EEA manufacturer you are representing and not the size of your own legal entity.
- Initiate a legal entity change (only representative changes)
- Include documents which clearly explain how you represent the new non-EU manufacturer
- Update the IUCLID files, in particular section 1.7 ‘Only Representative information'
- Submit a spontaneous update in the new legal entity to report the change in the dossier
You may need to pay a fee to complete the process. More information related to the outcome of a legal entity change can be found at Q&A 388.
As an only representative, you are fully responsible and liable for fulfilling all obligations of importers for the substances you are responsible for. These do not only pertain to registration but also to all other obligations of importers under REACH. As an only representative, you need to register the imported quantities depending on the contractual arrangements with the ‘non-EU manufacturer'.
You can represent one or several ‘non-EU manufacturers'. If you act on behalf of several ‘non-EU manufacturers', you must submit a separate registration for each of these manufacturers.
Your registration dossier should contain all uses of the importers covered by the registration. You need to keep an up-to-date list of importers within the same supply chain of the ‘non-EU manufacturer' and the tonnage covered for each of them, as well as information on the supply of the latest update of the safety data sheet.
For further information see chapter 2.1.2.5 "Only representative of a non-EU manufacturer" in the Guidance on registration:
If the non-EU manufacturer you represent is included in a list of legal entities targeted by sanctions or associated with anyone included in that list according to the relevant EU legislation on restrictive measures, you are advised to contact ECHA as soon as possible via the contact form. We will guide you in providing information related to the non-EU manufacturer, including information on the connection between the non-EU manufacturer and the sanctioned entity. Please note that, in any event, our advice will focus only on the implementation of legislation within ECHA’s remit, and not on your overall compliance with the restrictive measures.
For more information on the impact of the sanctions, please see the FAQs published by the European Commission at this link.
According to Article 2(9) of REACH polymers do not have to be registered. However, according to Article 6(3) of REACH, the reacted monomer substances and other substances that are incorporated in the polymers and that have not already been registered by an actor up the supply chain, are to be registered if both the following conditions are met:
- the polymer consists of 2 % weight by weight (w/w) or more of such monomer substance(s) or other substance(s) in the form of monomeric units and chemically bound substance(s);
- the total quantity of such monomer substance(s) or other substance(s) makes up 1 tonne or more per year (the total quantity in this context is the total quantity of the monomer substance(s) or the other substance(s) incorporated in the polymer).
The REACH Regulation defines polymers in Article 3(5) and monomers in Article 3(6). Monomer substances referred to in Article 6(3) relate only to reacted monomers which are incorporated in polymers.
The European Commission may according to Article 138(2) of the REACH Regulation present legislative proposals with requirements for the registration of polymers once a practicable and cost-effective way of selecting polymers for registration on the basis of sound technical and valid scientific criteria can be established.
Please note that in light of a recent decision of the ECHA Board of Appeal (Case A-001-2020), ECHA is still revising the Guidance for monomers and polymers
An impurity in a polymer is defined as an unintended constituent present in the manufactured polymer substance. It may originate from the starting materials, such as the monomers or any other reactants, or be the result of secondary or incomplete reactions during the production process. While it is present in the final substance it was not intentionally added. Examples of impurities in a polymer include unreacted monomers or other reactants, residual polymerisation catalyst, or any contaminant from the manufacturing process. The definition and detailed guidance on how to handle impurities can be found in Section 4.2.- 'Substances of well defined composition', Section 4.3.- 'UVCB substances', and Chapter 5- 'Criteria for checking if substances are the same' of the Guidance for identification and naming of substances Under REACH and CLP: http://echa.europa.eu/guidance-documents/guidance-on-reach
The provisions under the REACH Regulation with regard to information in the supply chain (Title IV), authorisation (Title VII), restrictions (Title VIII) and classification and labelling C&L (Title XI) may also apply to polymers. Further information on this issue is provided in Section 3.2.2- 'Application for authorisation', Section 3.2.3- 'Compliance with restrictions', 3.2.4- 'Classification and labelling', and Section 3.2.5- 'Information down the supply chain' of the Guidance for monomers and polymers: http://echa.europa.eu/guidance-documents/guidance-on-reach.
Natural polymers are understood as polymers, which are the result of a polymerisation process that has taken place in nature, independently of the extraction process with which they have been extracted (i.e. they may or may not meet the criteria set out in Article 3(39) of the REACH Regulation).
Following Article 2(9) of the REACH Regulation, any polymer meeting the criteria of Article 3(5) of the REACH Regulation does not have to be registered. This includes natural polymers, which are chemically modified (e.g. post-treatment of natural polymers).
Monomer substance(s) or other substance(s) in the form of monomeric units and chemically bound substance(s) originating from the natural polymer can for practical reasons be treated as "non-isolated intermediates" and do not have to be registered. The substances used to chemically modify the natural polymer and which are chemically bound within the final polymer need to be registered according to the REACH requirements.
This FAQ has been agreed by the Competent Authorities of the Member States (REACH CA) in October 2008.
Yes. The registration of a monomer or other substance chemically bound to a polymer has to include spectral data and a chromatogram of the original monomer or other substance used to manufacture the polymer.
If it is not technically possible, or if it does not appear scientifically necessary to include this information, the reasons need to be clearly stated. Generic spectral data or a generic chromatogram cannot be accepted as this would not reflect the actual composition of the monomer or other substance used to manufacture the polymer.
It may be the case that a company imports a type of polymer from different sources, and therefore a monomer or other substance used in the manufacture of this polymer probably also stems from different sources. Even when a company imports a polymer from just one source, it can happen that a monomer or other substance used in the manufacture of this polymer stems from different sources.
In these cases, the importer of the polymer is responsible for assessing the sameness of the monomer or other substance from the different sources. If they consider that the substances from the different sources are the same, they have to submit just one registration for this substance with one set of spectral data and one representative chromatogram. In this process, they might still have found out that the substance from the different sources has different impurity profiles. They need to then refer to these different compositions of the substance in their registration dossier.
Natural proteins may be considered as polymers under REACH provided that they have at least 50 weight percent of polymer molecules (in this case, molecules including a sequence of at least four amino acid monomer units) and the content of molecules presenting the same molecular weight remains below 50 weight percent.
Similarly, hydrolysed natural proteins may be considered as polymers if they fulfil the abovementioned criteria. If the degree of hydrolysis is to such an extent that less than 50 percent of the weight of the substance consists of polymer molecules (as defined in chapter 2.2 of the Guidance for monomers and polymers) and/or the amount of polymer molecules presenting the same molecular weight is at least 50 weight percent, the hydrolysed natural protein is not a polymer and, hence, is not covered by the registration exemption for polymers under REACH.
A polymer is a substance consisting of molecules characterised by the sequence of one or more types of monomer unit. Such molecules must be distributed over a range of molecular weights. Differences in the molecular weight are primarily attributable to differences in the number of monomer units.
Under REACH, a polymer is defined as a substance meeting the following criteria:
- Over 50 percent of the weight consists of molecules containing at least three monomer units which are covalently bound to at least one other monomer unit or other reactant; and,
- The amount of molecules presenting the same molecular weight must be less than 50 percent of the weight.
For further information see chapter 2.2.3.7 of the Guidance on registration:
Full details on polymers are available in the Guidance for monomers and polymers:
REACH imposes registration obligations only on manufacturers or importers (and, in specific cases, on producers or importers of articles). The registration obligation does not apply to downstream users or distributors. Therefore, the registration obligation does not apply to you if you have:
- manufactured or imported pre-registered substances before the registration deadline; and
- completely ceased such activities and only used the substances and/or supplied them after that.
For example, importers for whom the last batch of the substance was imported on 31 May 2018, at the latest, may continue to use and/or supply that substance after the deadline without a registration, without time-limitation. Since the pre-registered substance has not been registered, there is no obligation to notify ECHA of the cessation of manufacture/import. Article 50(2) and (3) of the REACH Regulation does not apply to this situation.
If you have not ceased your activities before the registration deadline, you have an obligation to register the substance according to Article 6 of REACH.
If you are a downstream user (or any other actor down the supply chain) who is not subject to the registration obligation, you can continue to use, without time-limitation, and/or supply quantities of the substance that were manufactured or imported before the registration deadline.
For the obligations of a downstream user or distributor and whether they are obliged to check the registration status of the substances on their own or in a mixture, please see Q&A 155
Substances that fulfil at least one of the following criteria may be considered as phase-in substances in accordance with Article 3(20) of the REACH Regulation:
- Substances listed in the European Inventory of Existing Commercial chemical Substances (EINECS);
- Substances that have been manufactured in the EU (including accession countries on 1 January 2007) but have not been placed on the EU market after 1 June 1992;
- Substances that qualify as a so-called ‘no-longer polymer’.
Detailed information can be found in the Guidance on registration, Section 2.3.1. – Phase-in substances vs non phase-in substances.
- Substance EINECS and CAS number (if available) and other identity codes;
- The first envisaged registration deadline;
- The names and other identifiers of related substances that pre-registrants have, i.e. those for which the available information may be relevant for performing adaptation of testing requirements using read across, (Q)SARs and/or grouping of substances.
Möglicherweise hat ein Prä-SIEF zahlreiche Mitglieder, von denen jedoch die meisten im Hinblick auf die Registrierung unentschlossen sind oder entschieden haben, keine Registrierung vorzunehmen. Der Status „Inactive“ (Inaktiv) im Prä-SIEF zeigt, dass das Unternehmen keine Registrierung vornehmen wird, allerdings wird er nicht von allen Unternehmen angegeben.
Antworten Unternehmen nicht auf Ihre E-Mails, könnten Sie in Betracht ziehen, sie auf anderem Wege zu kontaktieren (beispielsweise im Falle veralteter E-Mail-Adressen). Sie könnten sich dabei auf Unternehmen beschränken, die Sie kennen und bei denen Sie davon ausgehen, dass sie möglicherweise eine Registrierung vornehmen.
Darüber hinaus wäre es sinnvoll, regelmäßig die PDF-Datei mit den Vorregistranten herunterzuladen, um zu prüfen, ob sich die Kontaktdaten geändert haben. Bitte beachten Sie, dass Mitteilungen möglicherweise als Spam herausgefiltert werden.
Bleiben weitere Versuche der Kontaktaufnahme erfolglos, dokumentieren Sie die unternommenen Versuche und stellen Sie sich darauf ein, Ihren Stoff alleine zu registrieren. Teilen Sie Ihre Fortschritte den anderen Mitgliedern des Prä-SIEF per E-Mail mit. Ziehen Sie in Erwägung, als „SIEF Formation Facilitator“ (Vermittler der SIEF-Bildung, SFF) zu fungieren, sodass Sie im Prä-SIEF angeben können, wie sich andere mit Ihnen in Verbindung setzen können.
Handelt es sich bei Ihrem Stoff um einen komplexen Stoff, ziehen Sie die Möglichkeit in Betracht, dass andere Hersteller/Importeure des Stoffes andere Identifikatoren verwendet und sich einem anderen Prä-SIEF angeschlossen haben. Suchen Sie in der Liste der vorregistrierten Stoffe nach Prä-SIEFs, mit denen Sie sich möglicherweise zusammenschließen könnten.
The pre-registrations are not valid as of 1 January 2020 following the Commission Implementing Regulation (EU) 2019/1692.
Companies must register substances that they manufacture or import in quantities of more than one tonne per year in the EU/EEA before they start these activities, unless the substances are exempt from registration.
If you are adapting the information requirement based on existing data according to REACH Annex XI section 1, you should provide the applicable data (e.g. read-across study, Q(SAR)) as an endpoint study record, marked with the appropriate ‘Adequacy of study’ and ‘Type of information’. You should not report existing data as a data waiving record.
If you are waiving (omitting) the information requirement based on the provisions of Column 2 of the relevant requirement, or Sections 2 or 3 of Annex XI, you should include a data waiving record clearly documenting the justification for the waiving. Any available data that support this justification (e.g. a property of the substance, classification) should be reported in the relevant IUCLID section. For transparency, you may wish to link this information in the data waiving record using the ‘Cross-reference’ field.
For further information on how to complete endpoint study records in IUCLID, please refer to chapter 9.7 of the manual How to prepare registration and PPORD dossiers available on the ECHA website.
A: If the character limit in the free text field next to the value ‘other:’ does not allow you to provide sufficient information to justify the waiving, you can use the following IUCLID fields in the Administrative data part:
- ‘Remarks’ field below the picklist value ‘other:’
- ‘Justification for type of information’ – this field has a higher character limit and allows for a longer argumentation.
In addition, should you need to provide further information in the form of a report or schematic, you can use the field ‘Attached justification’ to attach the supporting document. To refer to information provided elsewhere in the dossier, you can also use the field ‘Cross-reference’.
However, it is important to note that the main argumentation for the data waiving has to be included in the fields ‘Other’, ‘Remarks’ or the field ‘Justification for type of information’ of the relevant endpoint study record. Only referring to somewhere else in the dossier (e.g. another section, an endpoint summary) or to an attachment is not a valid approach to justify the data waiving and such a record will not pass the completeness check.
At Annex X, the EOGRTS is the standard information requirement to address reproductive toxicity. If you would like to wait for the results of another test (e.g. results from a 90-day study or from a prenatal developmental toxicity study) before starting the EOGRTS, you should indicate this in IUCLID section 7.8.1 with an endpoint study record flagged as testing proposal.
To indicate an endpoint study record as a testing proposal for EOGRTS, set the field ‘Type of information’ to ‘experimental study planned’ and choose in the field ‘Endpoint’ one of the picklist values referring to the EOGRTS. In the testing proposal record you can explain the sequential testing and the test guideline(s) that you intend to follow. ECHA will evaluate this testing proposal and may set a timeline to allow this sequential testing.
At Annex IX, it is indicated in Column 1 of 8.7.3 that the extended one-generation reproductive toxicity study (EOGRTS) must be performed “if the available repeated dose toxicity studies (e.g. 28-day or 90-day studies, OECD 421 or 422 screening studies) indicate adverse effects on reproductive organs or tissues or reveal other concerns in relation with reproductive toxicity”. Therefore, the EOGRTS is not an automatic requirement at Annex IX, but relies on the indication of adverse effects from other tests.
If you are awaiting the results of such test(s) to determine if you need to conduct the EOGRTS, you can indicate this in an endpoint study record flagged as data waiving in IUCLID section 7.8.1. To indicate an endpoint study record as a data waiving for EOGRTS pending the outcome of another study, set the field ‘Data waiving’ to ‘study not scientifically necessary’ and choose in the field ‘Endpoint’ one of the picklist values referring to the EOGRTS. In the data waiving record, select under ‘Justification for data waiving’ the value ‘other:’ and explain that you are awaiting the outcome of another study. Be explicit on which study/studies you refer to, preferably by indicating the relevant section(s) in the field ‘Cross-reference’. If you have any ECHA/authority decision on the test, also include the decision number in the data waiving justification.
By following this approach, you do not need to submit a testing proposal for the EOGRTS at Annex IX, before you know if this study is needed. Once the repeated dose toxicity study is finalised, you should without delay submit an update of the registration, and in addition to reporting the results of the finalised test, in section 7.8.1 either (i) amend the data waiving record to explain that no adverse effects in line with Column 1 of 8.7.3 were observed in the test; or (ii) replace the data waiving record with a testing proposal for the EOGRTS.
REACH Annex X requires registrants to provide two pre-natal developmental toxicity studies in different species. For each of the two species, you need to provide an endpoint study record in IUCLID section 7.8.2 indicated as key study, weight of evidence, data waiving, or testing proposal.
In the situation you refer to, you have received the decision to carry out the PNDT test in two species, and you have started the testing in the first species. However, you would like to wait for the results of the PNDT test in the first species before initiating the second species study. In this situation, you should include in IUCLID section 7.8.2 the following endpoint study records:
First species record:
- You should indicate the endpoint study record for the ongoing first species PNDT test as a data waiving. Set the field ‘Data waiving’ to ‘other justification’. Under ‘Justification for data waiving’ select the value ‘other:’ and type the following sentence in the adjacent text field: “This information will be submitted later based on ECHA decision number TPE-F-xxxxxxxxxx-xx-xx”, where you replace the “x”-characters with the decision number issued to you by ECHA.
Second species record:
- You should indicate the endpoint study record for the second species PNDT test as a data waiving. Set the field ‘Data waiving’ to ‘other justification’. Under ‘Justification for data waiving’ select the value ‘other:’ and type the following sentence in the adjacent text field: “The second species PNDT test may be performed sequentially once the first species test results are available. This information will be submitted later based on ECHA decision number TPE-F-xxxxxxxxxx-xx-xx”, where you replace the “x”-characters with the decision number issued to you by ECHA.
Once a study is finalised, you must without delay submit an update of the registration, and report the results of the finalised test in IUCLID section 7.8.2.
Note that the only reasons for why the testing in the second species could be permanently waived based on the results of the first species study are listed in REACH Annex X, section 8.7.2, column 2.
For further information on the information requirements for the PNDT studies, please read the Newsletter, issued on October 2014.
REACH Annex X requires registrants to provide two pre-natal developmental toxicity studies in different species. For each of the two species, you need to provide an endpoint study record in IUCLID section 7.8.2 indicated as key study, weight of evidence, data waiving, or testing proposal. Before carrying out a test in vertebrate animals to fulfil information requirements for REACH Annexes IX and X, you need to first submit a testing proposal to ECHA and receive the decision to carry out the test.
In the situation you refer to, you are about to submit a testing proposal for the PNDT study at Annex X. You should therefore include in IUCLID section 7.8.2 the following endpoint study records:
First and second species records:
- You should create separate endpoint study records for the first and second species and indicate each of them as a testing proposal by setting the field ’Type of information’ to ‘experimental study planned’. Provide information on the ‘Guideline’, ‘Test material information’ and ‘Species’ of the planned test.
- In addition, ensure to include in the field ‘Justification for type of information’ of both records the considerations for why the adaptation possibilities offered by the REACH Regulation cannot be used to address the information requirement and animal testing is necessary.
- Finally, include in at least one of the records in the field ‘Attached justification’ the clarification of that you propose to carry out the testing of the two species in a sequential manner (depending on the results of the PNDT study in the first species).
Once a study is finalised, you must without delay submit an update of the registration, and report the results of the finalised test in IUCLID section 7.8.2.
Note that the only reasons for why the testing in the second species could be permanently waived based on the results of the first species study are listed in REACH Annex X, section 8.7.2, column 2.
For further information on the information requirements for the PNDT studies, please read the Newsletter, issued on October 2014.
- Substance is meant to present in the material matrix forming an article. Typical technical functions include plasticisers, fillers, flame retardants, pigments or stabilisers in plastic articles; dyes, finishing or sizing agents in textile, paper and leather; or alloying elements in metal articles.
- Substance is meant to be present in the surface layer protecting an article or delivering a certain appearance. Typical technical functions include binders (film formers), driers, pigments, plasticisers, stabilisers in the coatings for metal articles or wooden/mineral elements in building and construction.
- Substance is meant to be present on the article surface to promote or prevent adhesion to other surfaces. Typical technical functions include adhesion promoters, binders in adhesives, antiadhesives, release agents remaining on surfaces of mineral construction elements.
- Substance is meant to be present on article surface during longer periods of article service life, such as polishing, waxing or impregnation agents.
- Substance is a component in printing inks applied to packages or print media. Typical technical functions include binder, pigment, dyes.
- Substance is meant to be present in a plastic compound (or master-batch) in order protect the material against degradation by heat during processing (heat stabiliser).
- Anti-freeze agents and de-icers
- Cleaning and chelating agents
- Etching agents
- Flocculation or floatation agents
- Fuels and fuel additives
- Agents in hydraulic fluids, heat transfer fluids or other functional fluids
- Lubricating agents
- Solvents
- Within a use, report the technical functions that relate to the product(s) relevant for the use described.
- Functions excluding each other (e.g. flame retardant and fuel) should not be included in the same use.
- The technical function should be consistent with the selected environmental release categories (ERC), which systematically distinguish between inclusion and no-inclusion into/onto articles
- It should be also compatible with the indication in field ‘Subsequent service life relevant for this use’ of IUCLID.
If you report one or more uses in Section 3.5.6: Service Life of IUCLID, and the sub-stance you register meets the criteria of Article 14(4) of REACH for classification as haz-ardous (or considered PBT/vPvB), then your CSR must contain the corresponding expo-sure scenario(s). The exposure scenario(s) for service life are expected to address the conditions driving the release/ exposure. This usually includes the concentration of the substance in the material forming the article, the type of activity (associated with PROC) with the article and the related conditions of use for workers, or the type of article (asso-ciated with AC*) and related conditions of use for consumers.
*Note that for consumer activities, the use descriptors Article Category (AC) 1,2 and 3 refer to multi-material complex objects, which cannot be assessed as such in a contrib-uting scenario without further information on the type of material in which the substance occurs. Registrants are therefore advised to refer their contributing activities by consum-ers and their assessment to the material-based Article categories AC 4 to AC 13.
Each contributing scenario usually requires an estimate of the releases from the article and a corresponding exposure estimate and risk characterisation. Tools are available to help with this: ECHA's practical guide Describing uses of additives in plastic material for articles and estimating related exposure provides an overview on methods and tools that contains useful information also for other materials.
When considering how to complete your CSR to assess one or more uses (Service Life) reported in section 3.5.6 of IUCLID there may be situations that justify the absence of quantitative exposure assessment if relevant explanation is provided. The expected ex-planation depends on the situation you encounter:
- The substance is contained in concentration in the article material below the cut-off values as laid down in Regulation 1272/2008 (CLP) in relation to the mixture classification (using by analogy Article 14(2) of REACH, as a benchmark). If so, your dossier must contain explanation on the points listed under section A below.
- The substance reacts on use, and hence is not available for exposure anymore during service life. If so, your dossier must contain explanation on the points listed under section B below.
In contrast, simply stating that a substance is expected to be released from the article only in very small [negligible] amounts is not considered a valid argument to justify the absence of a quantitative assessment. The extent of exposure during handling or pro-cessing of articles depends on the conditions of use (e.g. abrasive processes, elevated temperature, intensity of dermal contact), the concentration of the substance in the arti-cle material and the interaction between the substance and the article material. In addi-tion, the hazard of the substance plays a role when determining whether the release is sufficiently low to guarantee control of risk. If situation A/B do not apply, you are there-fore expected to quantify the release, exposure and risk resulting from the service life use. If you don’t expect any reasonably quantifiable release on one or more routes, you may set the relevant release estimate(s) to zero and describe the related conditions of use in the contributing scenario. These conditions of use may, for example, include:
- no dermal contact, because the substance is embedded in the internal parts of the article, or the substance cannot migrate through a shielding surface layer.
- no oral contact during service life, because mouthing of the material by consumers or food contact not foreseen.
- no water contact during service life because the material is not foreseen to be used in articles for outdoor use, in pipes or similar articles, and also cleaning with water is not foreseen (e.g. as for flooring material).
A) Justification related to low concentration
To justify the absence of quantitative exposure assessment for certain service life uses due to the low concentration of your substance in the article material, your reasoning must include: the concentration of the substance in the article material and the following information:
- If the concentration is below 0.1% (this corresponds to the lowest concentration in table below), no further reasoning is expected at TCC, unless a specific con-centration limit (below 0.1%) for mixture classification is applicable to the sub-stance (in which case such cut off should be used). In addition, for substances classified for the environment as ‘Acute Category 1’ or ‘Chronic Category 1’, 0.1% is to be divided by the M-Factor to determine the substance-specific cut-off.
- If the concentration is above 0.1%, you must provide the hazard category(ies) and class(es) of your substance and the corresponding cut-off(s) from CLP, indi-cating the concentration above which your substance must be taken into account for the purposes of mixture classification. Based on this, you can demonstrate that the concentration in the article material is below the lowest cut-off relevant to your substance.
Such justification must be provided for each exposure scenario to which it applies.
B) Justification related to reaction on use
If a hazardous substance reacts on use, such reaction may be fast and complete (no re-sidual parent substance available for exposure) or some parent substance may remain, and the reaction product may be more hazardous or less hazardous compared to the parent substance. Therefore, the “disappearance” of the parent substance alone is not a sufficient argument for the absence of the exposure assessment.
To justify the absence of quantitative exposure assessment for certain service life uses you must quantify the residual concentration of the parent substance, and you must pro-vide information on the chemical nature and the known/expected hazard of the transfor-mation products. Your reasoning must include the following elements:
- To justify the absence of assessment of your substance:
- Provide an explanation how the substance transforms (including the reaction mecha-nism and the identity of the transformation products) and quantify the remaining re-sidual concentration of the substance in the article material (under the conditions of use of the service life).
- Compare the concentration of the substance in the article material with the cut-off concentration from the mixture classification rules (see table below) and provide the information as specified in point A above
- To justify the absence of assessment for the transformation product(s):
- Explain the available evidence that the transformation product(s) do(es) not meet the criteria for being considered hazardous.
Such justification should be provided for each exposure scenario to which it applies.
REACH-IT has been tested with:
- Google Chrome 48.0 and higher on a Microsoft Windows platform
- Internet Explorer 11.0 and higher on a Microsoft Windows platform
- Mozilla Firefox 44.0 and higher on a Microsoft Windows platform
The use of untested browsers may reduce REACH-IT functionalities and cause application errors in REACH-IT. Using an older version of the browsers, or different software platform, may cause incompatibility issues with REACH-IT functionalities. Therefore, users are advised to upgrade their internet browser as mentioned above.
There is no need to synchronise the company details in REACH-IT and IUCLID. The legal entity information is always extracted from the information in REACH-IT.
However, if you wish to do so, we recommend updating the information in REACH-IT, and later exporting it to your IUCLID 6. If you update your LEO information in your IUCLID 6 application, remember also to update your LEO information in REACH-IT.
For more information, see the manual How to prepare registration and PPORD dossiers, chapter 2.1, How to update and synchronise the LEO information.
REACH-IT and IUCLID support the following file types: jpg, jpeg, pdf, doc, docx, rtf, txt, tiff, mol, png, gif, xls and xlsx.
The EC number can be downloaded from REACH-IT either from the Joint submission Object, the Inquiry or the Pre-SIEF page.
- Log in to REACH-IT, go to the relevant submission page and click on the button ‘Export assigned EC number or Export EC substance’, as shown below:

- Once the assigned EC number has been exported, go back to the IUCLID home page and click on ‘Import’ as shown below.

- Once the assigned EC number has been imported go back to the IUCLID home page and click on ‘Reference substance’ as shown below.

- Click on the button ‘New reference substance’ as shown below.

- ‘Add’ the newly uploaded reference number to the reference number inventory by putting the name of the reference substance and click create and open.

- Fill in the reference substance identity information and save the entry.
After you have submitted your registration dossier to ECHA you may realise that you made a mistake during its preparation. This might be the case, for example, if you accidentally introduced faulty information in the dossier (e.g. incorrect information in one of the study summaries) and noticed this only after you submitted the dossier to ECHA. In this case you should without undue delay submit the amended dossier as a spontaneous dossier update via REACH-IT, indicating in the dossier header the reason(s) why you are spontaneously updating it as well as the references of the previous valid submission (i.e. the "last submission number"). Such an update would not be subject to a fee, unless the mistake is related to a chargeable element such as an increase in tonnage band, or confidentiality claims for information listed in Article 119(2).
Some mistakes may prevent a successful submission of the dossier to ECHA. These are failures in the business rules or the completeness check. If your submission fails either of these checks, you will be alerted to the failure in the form of a task in REACH-IT, which prompts you to amend the dossier.
Following the entry into force of the ‘Commission Implementing Regulation (EU) 2016/9 on Joint Submission of Data and Data Sharing', new checks were put in place to ensure that all submissions are in line with the ‘one substance, one registration' (OSOR) principle. According to the OSOR principle there can only be one joint registration per substance per registration type (full or intermediate).
Furthermore, if an active joint submission already exists (i.e. the lead registrant has successfully submitted the lead dossier), individual registrants (i.e. not part of the joint submission) for the same registration type (full or intermediate) are blocked from updating their dossiers, unless they become members of the joint submission.
ECHA advises you to take the following steps:
- Find your co-registrants.
- Establish substance sameness and negotiate on data sharing.
- For the scenarios described in Q&A 1171, you can partially or fully opt-out from information submitted jointly by your co-registrants. However, your submission still has to be in the framework of the joint submission. Therefore, certain administrative fees may be applicable towards the joint submission.
- If you cannot agree on the data and cost sharing, you can file a data-sharing dispute with ECHA. The dispute mechanism should be used as a last resort and you will need to demonstrate that you have made every effort to reach a voluntary agreement.
- If you wish to submit a partial or full opt-out, but cannot come to a voluntary agreement with the lead registrant on the conditions for joining the joint submission, you can also use the above-mentioned dispute mechanism.

Business rule 027 (BR027) failure occurs if the substance identifier in your IUCLID dossier does not match the substance identifier on the REACH-IT joint submission page. To avoid BR027 failure in your future submissions, see the following check list and carry out the steps that apply to your case.
- Check that you have selected the correct joint submission in REACH-IT when submitting your IUCLID dossier.
- Check that the EC number in your IUCLID dossier is the same as the EC number of the joint submission in REACH-IT. You can export the assigned EC number either from the REACH-IT joint submission page, from the Pre-SIEF page, or from the inquiry submission report page. Import the EC number into IUCLID and assign it to your reference substance, as specified in Q&A 1258.
- If no EC number exists for your substance yet, check that the spelling of your substance name on the REACH-IT joint submission page is identical to the spelling in your IUCLID dossier section 1.1 IUPAC name. Pay attention to additional spaces and special characters.
- If your substance is a multi-constituent substance (e.g. a reaction mass of…), make sure that all the constituents indicated in section 1.2 of IUCLID are identical to the constituents included in the REACH-IT joint submission page.
- If you are a lead registrant of a new joint submission for a substance of Unknown or Variable composition, Complex reaction products or Biological origin (UVCB) with no valid pre-registration or inquiry number, and have tried the above mentioned fixes, please contact ECHA using the ECHA contact form to receive further instructions on how to successfully submit a registration.
While creating your dossier, ECHA advises you to use the IUCLID Validation assistant plug-in to help you to detect any business rules and technical completeness check failures in your dataset and dossier, as explained in Q&A 0392. Please address any detected failures before re-submitting your dossier update to ECHA through REACH-IT.

Business rule 034 (BR034) failure occurs when you submit a spontaneous update instead of requested update. When your previous submission fails the technical completeness check (TCC), you are expected to submit a requested update. A communication regarding the TCC failure is sent to you via REACH-IT. You also receive an update request under ‘Tasks’ in your REACH-IT account with instructions on how correct the TCC and submit a requested update.
When you create your dossier update, you must tick the boxes 'The submission is an update' and 'Further to a request/decision from a regulatory body’.
Add your last submission number, in this case, the submission that failed the technical completeness check. In the box below, enter the communication number in the ‘Number’ field, as shown in the screenshot.

You can find the submission number and the communication number in REACH-IT under ‘Tasks’ and also under ‘Key documents’.
While creating your dossier, we also advise you to use the IUCLID Validation assistant plug-in to help you to detect business rules and technical completeness check failures in your dataset and dossier, as explained in Q&A 0392. Please address any detected failures before re-submitting your dossier update to ECHA through REACH-IT.

Business rule 35 (BR035) failure occurred because the last submission number you have indicated in your dossier is incorrect. To address the failure, you will need to indicate the last successful submission number for the particular substance in the dossier header in IUCLID, and then re-submit the dossier in REACH-IT.
You can find your previous successful submission number in REACH-IT by going to ‘Substances’ and searching for your registration using the substance name or EC number. The submission number is presented in the results page, as shown in the screenshot below (1).
If you are expected to submit an update due to technical completeness check failure, you need to use the ‘In progress’ submission number (2).
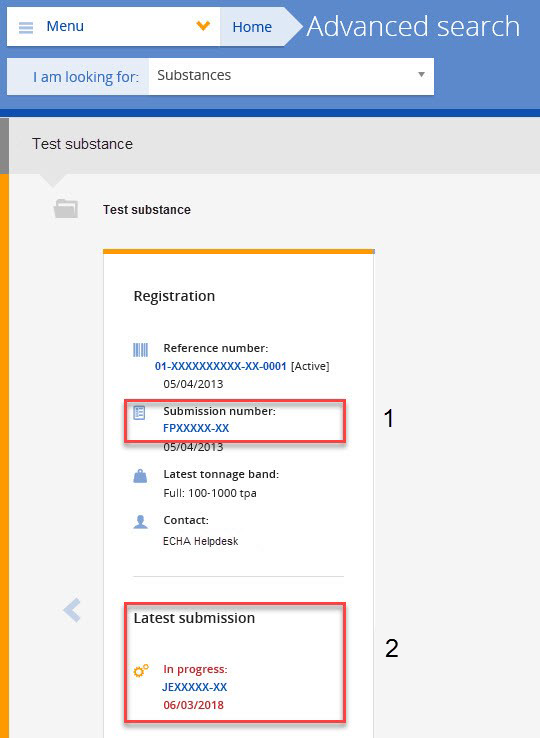
Assign the correct submission number in the IUCLID dossier creation wizard by ticking the box ‘The submission is an update’ and insert the last submission number, as shown in the screenshot.
While creating your dossier, we also advise you to use the IUCLID Validation assistant plug-in to help you to detect business rules and technical completeness check failures in your dataset and dossier, as explained in Q&A 0392. Please address any detected failures before re-submitting your dossier update to ECHA through REACH-IT.

Your submission has failed business rule 038 (BR038) because you already have a successful submission for the substance and you are expected to submit an update.
To address the failure, you need to assign the last successful submission number of the substance you are submitting the update for in the IUCLID dossier header. Indicate the reason for the update by choosing the appropriate update type, ‘Spontaneous update’ or ‘Further to a request/decision from a regulatory body’.
You can find your last successful submission number in REACH-IT by going to ‘Substances’ and searching for your registration using the substance name or EC number. The last successful submission number is listed on the results page, as shown in the screenshot below.

For a spontaneous update, carry out the following steps, as shown in the screenshot below (Desktop view and IUCLID Cloud view).
- Assign the submission number in IUCLID dossier creation wizard by ticking the box ‘The submission is an update’ and inserting the last successful submission number that corresponds to the substance.
- Select the type of update, ‘Spontaneous update’.
- Select the appropriate justification for the update. Create a block by clicking the button "New item" and make a selection from the drop down list. If you select ‘other’, you are required to give the reason in the adjacent free text field.

For a requested update, refer to the assessment outcome communication sent to you via REACH-IT. You can find the communication on the submission page under the ‘Key documents’ tab. The communication provides full details on what information is missing from the dossier and a deadline by when you should submit this information.
For a requested update, carry out the following steps, as shown in the screenshot below (Desktop view and IUCLID Cloud view).
- Assign the latest submission number in IUCLID dossier creation wizard by ticking the box ‘The submission is an update’ and inserting the last successful submission number, which corresponds to this substance.
- Select the type of update, ‘Further to a request/decision from a regulatory body’.
- Create a block by clicking the button "New item" and insert the annotation number that corresponds to the submission for which further information was requested (e.g. assessment outcome communication number XXX-X-0000000000-00-00/X). In the ‘Remarks’ field, enter your remarks as free text.

While creating your dossier, ECHA advises you to use the IUCLID Validation assistant plug-in to help you to detect any business rules and technical completeness check failures in your dataset and dossier, as explained in Q&A 0392. Please address any detected failures before re-submitting your dossier update to ECHA through REACH-IT.

Your submission has failed business rule 053 (BR053) because the EC/list number in section ‘1.1 Identification’ your IUCLID dossier does not match the EC/list number of the inquiry indicated in section ‘1.3 Identifiers’.
To address the failure, you need to ensure that:
- the substance identification in section ‘1.1 Identification’ of the IUCLID dossier corresponds to the substance identification associated to the inquiry number indicated in section ‘1.3 Identifiers’;
- the inquiry number indicated in section ‘1.3 Identifiers’ has been copied/typed in the correct format.
To select the EC/list number provided by ECHA as a result of your inquiry assessment, you need to export this EC number from REACH-IT in .i6z format into IUCLID. For instructions on how to export the EC number from REACH-IT to IUCLID, see Q&A 1258.
You can find your inquiry number in REACH-IT by going to ‘Substances’ and searching for your inquiry using the substance name or EC number. The inquiry number (e.g. 06-0000000000-00-0000) will be listed on the results page.
While creating your dossier, ECHA advises you to use the IUCLID Validation assistant plug-in to help you to detect any business rules and technical completeness check failures in your dataset and dossier, as explained in Q&A 0392. Please address any detected failures before re-submitting your registration dossier to ECHA via REACH-IT.

Business rule 119 (BR119) failure occurs when the lead registrant indicates in their dossier update that they will not provide the optional information (i.e. chemical safety report (CSR) and guidance on safe use (GoSU)) on behalf of its members although in the previous submission they had indicated that they would provide this information.
The following scenarios may trigger BR119 failure:

If the lead registrant initially indicated that they would provide the CSR and/or GoSU on behalf of all members of the joint submission but no longer wishes to do so, they should contact ECHA using the ECHA contact form and request the modification of the joint submission coverage. Before contacting ECHA:
- The lead registrant should first ensure that the changes will not affect any members of the joint submission.
- Members relying on the CSR and/or GoSU provided by the lead registrant need to submit a spontaneous update indicating that they will provide individual CSRs and/or GoSUs.
Once ECHA has performed the changes, the lead registrant will need to submit a spontaneous update.
When submitting the spontaneous update, member or lead registrants should indicate in the IUCLID dossier the submission type as ‘spontaneous update’, the ‘Justification’ select the reason ‘New or update of CSR or guidance of safe use’ as well as entering a clarification in the ‘Remarks’ text box.
While creating the dossier, we advise you to use the IUCLID Validation assistant plug-in to help you to detect business rules and technical completeness check failures in your dataset and dossier. For further instructions on how to use the validation assistant please refer to Q&A 392.

Business rule 130 (BR130) failure occurs when a lead registrant submits a dossier update in which the joint submission tonnage band indicated is lower than the joint submission tonnage band for the previous submission. The tonnage band of the joint submission is derived from the submission type (IUCLID template) chosen for the substance when the lead dossier was first created.

The individual tonnage band(s) specific to the lead registrant can be indicated in the dossier header.

If the lead registrant wishes to lower their individual tonnage band, they can refer to Q&A 1300 for further instructions.
If the lead registrant wishes to lower the tonnage band of their joint submission, they can refer to the Q&A 1299 for further instructions.
While creating your dossier, ECHA advises you to use the IUCLID Validation assistant plug-in to help you to detect any business rules and technical completeness check failures in your dataset and dossier, as explained in Q&A 0392. Please address any detected failures before re-submitting your registration dossier to ECHA via REACH-IT.
- Save the downloaded file locally on your computer.
- Open Excel. In the “Data” tab click on "Get Data -> From File From Text/CSV" to open a wizard.
- Go to the location of the CSV file and import it.
- The wizard will open. Set the “File Origin” (character encoding) to “65001: Unicode (UTF-8)” from the dropdown list.

- Set the “Delimiter” type to “Semicolon” from the dropdown list.

- Verify that the example data are correct and select “Load”.

- In case you need to make any changes, select the “Transform Data” option.
A substance must be registered before the manufacture or import can start at an annual quantity of one tonne or more. Before the registration of such substances, the manufacturer or importer has to make an inquiry to ECHA regarding any previous registration for the same substance.
Upon registration, the registrant may have to wait for three weeks after the registration dossier is submitted before starting or continuing the manufacture or import of the substance (Article 21 of the REACH Regulation).
You can cease manufacture due to the following scenarios:
- You ceased manufacture/import due to commercial reasons according to Article 50(2) of the REACH Regulation. The registered volume in your registration is updated to zero and the status of your registration is changed to "inactive". If you restart manufacture/import of the substance, you should inform ECHA via the 'Restart manufacture or import' button available on the reference number page in REACH-IT.
- You ceased manufacture following a draft decision according to Article 50(3) of the REACH Regulation. The status of your registration will be changed to "Invalid". A new registration must be submitted if you wish to restart manufacturing/importing your substance.
Below are the steps on how to cease/restart manufacture in REACH-IT.

You should use the functionality ‘restart manufacture or import’ to reactivate the reference number, when a substance is manufactured/imported again.
Your registration remains your responsibility, so keep it always up-to-date.
Safety Data Sheet
If you need to give your customers safety data sheets, you will need to include your registration number in the safety data sheets the next time you update them. In addition, if you have conducted a chemical safety assessment and needed to develop exposure scenarios, you have to attach them to the safety data sheet, outlining the use-specific conditions of safe use of the substance. This needs to be done as soon as possible.
SME verification
If you have claimed you are an SME and have benefited from the reduced registration fees, ECHA may verify the size of your company. Make sure that your REACH-IT account includes all the necessary documentation to justify the size of your company.
Joint Submission management and Data sharing
We strongly recommended that you put in place a mechanism discuss within your joint submission and for dossier update, when necessary. For example, you must also share data and give access to the joint submission to newcomers. You can charge for access to the data by sharing the costs in a fair, transparent and non-discriminatory way.
Dossier evaluation
ECHA will check the compliance of at least 20% of the received registration dossiers to verify that the information submitted is compliant with the legal requirements. The outcome of this dossier evaluation may be a request for additional information. Additionally, ECHA will examine the testing proposals included in the dossiers. This will happen by 1 June 2022 for registrations submitted for the deadline of 31 May 2018, and within 180 days for other registrations.
ECHA will communicate with you via REACH-IT, if the information of your registration dossier is being assessed, so we invite you to check your account regularly.
No. The importer is the natural or legal person established within the European Union (EU, for REACH and CLP also covering the European Economic Area, EEA) who is responsible for import, i.e. the physical introduction (of goods) into the customs territory of the EU (Article 3(10) of REACH).
The responsibility for import depends on many factors such as who orders, who pays, and who deals with the customs formalities or the "INCOTERMS" chosen, but these might not be conclusive on their own. In many instances, the end receiver of the goods (the consignee) will also be the legal entity responsible for the import. However, this is not always the case.
If, for example, you order goods from a company (Company B) established in the EU, which acts as a distributor, you probably do not know where the goods originate from. Company B may choose to order the goods from either an EU-based manufacturer or from a non-EU-based manufacturer.
If the Company B chooses to order from a non-EU-based manufacturer, the goods may be delivered directly from them to you to save on transportation costs. If this happens, your company will be stated as the consignee on the simplified administrative document form and customs handling will take place in your country. Payment for the goods will, however, be settled between you and Company B.
Company B should be considered the legal entity responsible for the physical introduction of the goods into the customs territory of the EEA (importer). You would then be a downstream user. The obligation to register would consequently lie with Company B.
You, on the other hand, will have to be able to prove through documentation to the enforcement authorities that you are a downstream user, for example, by showing that the order was placed to the distributor.
In addition, when interpreting the term "importer" according to the REACH Regulation, it is not possible to fall back upon the Community Customs Code (Regulation (EEC) No. 2913/92), recast in Regulation (EU) 952/2013.
Only a natural or legal person established within the European Economic Area (EEA)/EU can be a registrant. Registration must take place when this person:
1. manufactures a substance within the EU in quantities of 1 tonne or more per year;
2. imports a substance into the EU of quantities of 1 tonne or more per year; or
3. has been appointed as an only representative according to Article 8 of REACH (see FAQs on Only Representative of non-EU manufacturer).
The national law of each country provides the specific provisions concerning natural or legal personality and when such a natural or legal person is established in its territory.
A company not established within the EU does not have direct obligations under REACH. For obligations of a non-EU company, please see to Q&A 12.
You can use the Navigator tool to determine your obligations under REACH and find the appropriate guidance.
A toll manufacturer is normally understood to be a company that manufactures a substance (on its own, in a mixture or in an article) in its own technical facilities following the instructions of a third party in exchange for an economic compensation. The substance is generally put on the market by the third party. This construction is, for example, used for an intermediate step in the production process for which sophisticated equipment is needed (distillation, centrifugation etc.). According to the REACH Regulation, manufacturers of substances are required to register the substances they manufacture above one tonne per year. From this point of view the toll manufacturer is a manufacturer and has to register the substance.
This scenario is further discussed in the factsheet Toll manufacturer under the REACH Regulation.
International companies sometimes have several daughter companies in the EEA/EU, often spread over several countries. If these subsidiaries of the parent company are separate legal entities from it, (a natural or legal person as defined under applicable national law), then each of those must determine if they qualify as registrant under REACH.
Please see Q&A 27 on who has to register a substance.
No. The obligation to register a substance applies only to actors established within the EEA. Thus, the registration of substances imported into the EEA on their own, in mixtures or, in certain cases, in articles will have to be done by the importer established in the EEA. This implies that each individual importer needs to register the substance. However, according to Article 8(1) of the REACH Regulation manufacturers of substances, formulators of mixtures or producers of articles established outside the EEA, can nominate an only representative established within the EEA to carry out the required registration. This will relieve the individual EEA importers within the supply chain of that non-EEA manufacturer from their registration obligations for these substances. They will be regarded as downstream users of this only representative. However, the registration obligation may still apply if the EEA-importers import the same substance from other non-EEA manufacturers.
More information on the only representative role can be found in Only Representative of non-EU manufacturer and in section 2.1.2.5-'Only representative of a "non-EU manufacturer"' of the Guidance on registration.
For more information, see: http://echa.europa.eu/contact/helpdesk-contact-form/enquiry-on-reach-from-non-eu-countries.
Yes. Companies are free to register a substance for a tonnage band which is above the actual tonnage of the substance. This is also reflected in Section 2.2.6.3- 'Calculation of the total volume' of the Guidance on registration.
A registration at a higher tonnage band will trigger a higher registration fee in accordance with Regulation (EC) No 340/2008. In addition, the technical dossier will need to comprise all the information required for the registered tonnage band. Practical advice on how to complete a IUCLID dossier is provided in the Manual How to prepare registration and PPORD dossier at: http://echa.europa.eu/manuals.
Each registrant has to calculate the yearly tonnage for the registration dossier. The yearly tonnage is calculated as the volume per manufacturer/importer per calendar year.
A registrant needs to update their registration without undue delay as soon as the ‘annual or total quantities’ they manufacture or import reach the next tonnage band threshold, as required by Article 22(1)(c). As soon as the annual volume of a substance that has already been registered reaches the next tonnage threshold, the manufacturer or importer has the duty to inform ECHA of the additional data required, following Article 12(2).
The list numbers published by ECHA are not official EC entries. However, we recommend you to assign such an entry to the substance you intend to register provided this list number is linked to a CAS number (list numbers starting with 6 or 8) or a chemical name (list numbers starting with 9) corresponding to a correct and specific identifier for your substance. If this entry is too generic for your substance and there is no appropriate EC entry available, you should not assign any list/EC number to your substance.
If your registration dossier fails the technical completeness check (TCC) for the first time, you are given a deadline of 4 months during which you have one chance to amend the incompleteness. If you fail to submit the complete dossier by this given deadline, your submission will be rejected.
Rejection of a new registration means that the registration number is not assigned to your substance and any fees paid for this registration will not be refunded or otherwise credited. Consequently, you may only start to manufacture/import the substance or to produce or import an article once you have a complete registration and ECHA has issued you a registration number. If you wish to proceed with your registration you need to make a new initial submission. It will be subject to a new completeness check and registration fee.
Rejection of a registration update means that you maintain your existing registration number but any new information included in that update will not be included in the ECHA’s database. Any fees paid for this registration update will not be refunded or otherwise credited. If the update covered a change of tonnage band, you may not manufacture/import the substance or produce or import an article containing it at the increased tonnage band until ECHA has confirmed that your registration update is complete.
If the update of the registration dossier was submitted to include nanoforms of the substance then the rejection means that, even though you maintain your existing registration number for the non-nanoforms of the substance, the registration does not cover the manufacture or import of the nanoforms of the substance. Registrants that manufacture/import nanoforms of the substance after 1 January 2020 without a registration that covers these nanoforms are in breach of the REACH regulation which may trigger enforcement measures. Therefore, if your registration update to include nanoforms was rejected, you need to, at earliest possible, submit a new registration update in order to fulfil your obligations for nanoforms.
As a first step, prepare your registration/PPORD dataset and dossier according to the advice given in the manual ‘How to prepare registration and PPORD dossiers’ available at: http://echa.europa.eu/manuals. Annexes 1-3 of the manual also give an overview of the business rules and technical completeness check rules that apply to registration and PPORD dossiers.
Next, use the IUCLID Validation assistant plug-in to help you detect business rules and technical completeness check failures present in your dataset and dossier. To run the Validation assistant, right click on your dataset in the Navigation panel → Validate → follow the steps in the wizard. The same validation should be performed on the dossier to make sure that no failures have been introduced during dossier creation.

While the Validation assistant cannot replicate all the checks performed by ECHA, it simulates the majority of the verifications done and helps you minimise the chance of failure during submission. It is important to keep in mind, that any failure in the Validation assistant Submission checks tab, that is left uncorrected before submitting your dossier in REACH-IT, will lead to your submission not being accepted by ECHA.
You can find a video tutorials on the use of the IUCLID Validation assistant in the link below:
https://www.youtube.com/watch?v=zQEncCL8cCE&index=4&list=PLOPGDACSd6qyDkdXwPua1Fjb5bJksY75k
Please note that if the Validation assistant does not indicate any failures, this is not an automatic confirmation of that your dossier is complete. As of 21 June 2016, the technical completeness check includes additional manual verifications of the registration dossier by ECHA staff. These checks cannot be replicated using the Validation assistant plug-in; the related completeness issues cannot be displayed by the tool.
Information on the areas of the additional verifications can be found in the following location; https://echa.europa.eu/documents/10162/13652/manual_completeness_check_en.pdf.
We also recommend you to have a look at our webinars on the completeness check process:
https://echa.europa.eu/-/revised-completeness-check-what-changes-and-how-you-can-prepa-1
If your dossier is submitted before the compliance check deadline has passed, you may follow the deadline given in the TCC letter. ECHA will not continue with the Evaluation process before you have submitted the requested update for the technical completeness check failure(s).
If you fail TCC for the second time while submitting the requested update, your submission will be rejected.
If at this point the compliance check deadline has already passed, it means that ECHA has not received a response to the compliance check decision and may proceed with further regulatory actions. It is therefore advisable not to leave the submission of updates to regulatory requests to the last moment.
You can inform ECHA about not meeting the compliance check deadline via the contact form: https://echa.europa.eu/contact
There is no need to change the data waiving justification from free text to an available picklist phrase. A free text justification will be considered equally complete to a picklist value, if it is in line with column 2 of REACH Annexes VII-X, or sections 2-3 of Annex XI.
However, when preparing a dossier in IUCLID 6 from IUCLID 5 data, please review the selection in the field ‘Endpoint’ in particular for IUCLID sections where different information requirements can be addressed (e.g. 4.13, 7.8.1) to ensure that you have clearly indicated that the appropriate requirement is being waived.
The quality rules warn the user of common inconsistencies and shortcomings in the dossier. These warnings will not prevent you from successfully submitting your dossier in REACH-IT. However, leaving quality warnings uncorrected may lead to future clarification requests by ECHA.
The manual verification applies to all registration dossiers submitted to ECHA. The manual checks focus on ensuring that registrants who waive or deviate from the standard information requirements provide justifications foreseen by the legislation. Therefore, the extent of the verification depends on whether the dossier contains waiving of standard information or deviations from substance identification conventions, as well as on the specific requirements that apply to the registration type (i.e. lead, member, individual registration) and the registration scope (tonnage band, isolated intermediate).
For further information, please refer to the document ‘Information on manual verification at completeness check’ available at: https://echa.europa.eu/manuals.
In the task you have a link to the “Submission page” of the submission. Follow this link and go to the “Key documents” section at the bottom of the page. Here you can download the technical completeness check communication which contains all the relevant information regarding the failure, as well as the steps required by you.
The completeness check is based on REACH Article 20(2). It applies to all registration dossiers submitted to ECHA, (i.e. initial submissions and updates). The completeness check consists of two parallel verifications: (i) the technical completeness check, in which ECHA verifies that all the required elements have been provided in the registration dossier; and (ii) the financial completeness check, in which ECHA verifies that the registration fee has been paid, if applicable to that submission.
If during the technical completeness check the registration dossier you submitted is found incomplete, ECHA will issue a deadline by which you have only one attempt to submit the complete information. If you do not provide the requested information by the set deadline in the form of an update dossier, the submission will be rejected. In such a case, any registration fee you had paid in relation with this submission will not be refunded or otherwise credited.
You can find more information about the registration process on "From sbmission to decision" .
We also recommend you to have a look at our webinar on the technical completeness check process.
In general, testing for human health endpoints can be adapted (‘waived'), if the substance is used in the EEA exclusively in cosmetic products falling within the scope of the Cosmetics Regulation, and if the testing would not be necessary to fulfil the REACH requirements for the assessment of worker exposure.
Two main scenarios are foreseen where cosmetics-based waiving could be applied.
- In cases where imported products fall within the scope of the Cosmetics Regulation (EC No 1223/2009) and which, from the time of import, are neither further processed nor repackaged inside the EEA, an adaptation of animal testing requirements for human health endpoints can be sought, based on the absence of relevant worker exposure;
- In other cases, you may be able to seek an adaptation of an information requirement by demonstrating that the substance is handled under strictly controlled conditions during all stages of the life-cycle, other than the use as a cosmetic product (i.e. manufacture, formulation and/or packaging stage).
In all circumstances, you shall provide a reasoned justification for requesting the waiver.
Substances which have been registered, exported and then re-imported are exempted from registration under certain conditions.
To benefit from this exemption, you need to document that the following conditions are fulfilled:
- The substance must have been registered before it was exported from the EU.
- The substance already registered and exported must be the same, as the substance being re-imported.
- The substance must not only be the same but it must actually proceed from the same supply chain in which the substance was registered.
- The re-importer must have been provided with information on the exported substance as required by REACH (e.g. safety data sheet).
For further information, see the Guidance on registration, chapter 2.2.3.6 "Re-imported substance".
When a substance is used in food for humans or feedingstuffs for animals in accordance with the Food Safety Regulation ((EC) No 178/2002), the substance does not have to be registered.
This includes the use of the substance:
- as a food additive in foodstuffs (Council Directive 89/107/ECC);
- as a flavouring in foodstuffs (Council Directive 88/388/ECC and Commission Decision 1999/217/EC);
- as an additive in feedingstuffs (Regulation (EC) No 1831/2003);
- in animal nutrition (Council Directive 82/471/EEC).
Amounts of the same substance used for other uses than food and feedingstuffs are not exempted from registration. Only the amounts of the substance used in food and feedingstuffs are exempted from the registration obligation under REACH.
For further information, see the Guidance on registration, chapter 2.2.3.1.
The registration requirement for substances in articles (as required by Article 7 (1) of REACH) applies only if all the following conditions are met:
- the substance is intended to be released during normal and reasonable foreseeable conditions of use; and
- the total amount of the substance present in the article exceeds one tonne per producer or importer per year; and
- the substance has not yet been registered for that specific use. Pre-registrations, however, do not relieve you from the obligation to register.
If the substance has already been registered for your specific use, then you do not need to register. This registration can be done in your supply chain or any other supply chain. Chapter 3.3.1 of the Guidance on requirements for substances in articles provides advice on how to find out if the substance is already registered for that use.
In order to determine what is an article in this context, especially regarding border line cases between containers and articles, you can consult the Guidance on requirements for substances in articles
Substances that meet the criteria outlined in Article 57 of the REACH Regulation are commonly referred to as substances of very high concern (SVHCs). You must notify SVHCs present in articles (Article 7(2)) if the following conditions are met:
- the substance has been included in the Candidate List of SVHCs for authorisation; and
- the substance is present in articles above a concentration of 0.1% weight by weight (w/w); and
- the total amount of the substance in those articles (i.e. those containing more than 0.1% (w/w) of the Candidate List substance) exceeds one tonne per producer or importer per year; and
- exemptions do not apply.
Two specific exemptions can apply to the notification of substance in articles:
(a) exemption based on exclusion of exposure to humans or the environment during normal or reasonably foreseeable conditions of use and disposal (Article 7(3)) and
(b) exemption for substances already registered for that use (Article 7(6)).
You must notify Candidate List substances in articles at the latest six months after the SVHC has been included on the Candidate List for authorisation (Article 7(7)).
The Candidate List will be updated continuously when substances that meet the criteria of Article 57 are identified.
You can find further information at: http://echa.europa.eu/regulations/reach/candidate-list-substances-in-articles/notification-of-substances-in-articles.
The Candidate List can be found at: http://echa.europa.eu/candidate-list-table.
A substance is intended to be released from articles if it fulfils an accessory function which would not be achieved if the substance was not released. A scented children's toy, for example, is an article with intended release of substances, because fragrance substances contained in the toy are released in order to fulfil an accessory function, namely to scent. Consequently, the release of a substance because of ageing of articles, because of wear and tear or as an unavoidable side-effect of the functioning of the article, is generally not considered as an intended release, as the release as such does not provide a function in itself.
An intended release of a substance from an article has furthermore to occur under normal or reasonably foreseeable conditions of use. This means that the substance release has to occur during the service life of the article. Hence, a substance release during the production or disposal phase of the article's life cycle is not an intended release. Similarly, a release in an accident or due to any form of misuse which is not in accordance with the use instructions of the article, does not occur under normal or reasonably foreseeable conditions of use and is therefore not considered to be an intended release.
If the production/import ended before the SVHC was included in the Candidate List or before the notification obligation starts to apply (i.e. 6 months after a substance has been included in the Candidate List or 1 June 2011 for substances placed on the Candidate List before 1 December 2010) then you do not have to notify. However, you may still have obligation, under Article 33 of REACH, to provide the recipient of the article, or the consumer upon request, with sufficient information to allow safe use of the article, including, as a minimum, the name of that substance.
A permanent magnet should be considered as a substance or a mixture (and not an article) under REACH. This is because its shape, surface or design are less relevant for its function than its chemical composition. As a substance or a mixture, a permanent magnet is subject to the applicable provisions of the REACH and CLP regulations (e.g. it needs to be appropriately packaged and labelled).
Permanent magnets are used in different sizes and forms. They should create a (strong) permanent magnetic field and be stable to perform their main function of attracting or repelling other magnetic objects through a magnetic force (e.g. in cupboards to keep a door closed). They should also have high magnetic coercivity (i.e. they should be difficult to demagnetise).
The materials to be used to produce permanent magnets should either be either materials with permanent magnetic fields or materials with a susceptibility to be magnetised by applying an external magnetic field. The latter should also retain the imprinted magnetic pattern (high magnetic coercivity).
The magnetism is one example of a physical property that results from the chemistry of the materials an object is made of, given in the Guidance on requirements for substances in articles (subchapter 2.2). The Guidance also states that such material characteristics or properties are not to be confused with the shape, surface and design of an object.
The stability (magnetic coercivity) and the strength of the created permanent magnetic field appear to be the most important properties of a permanent magnet. Therefore, the magnetic properties of the permanent magnet, which are strongly related to its chemical composition, determine its function.
If you are a supplier of an article containing a substance, you have to provide to the recipient of the article (Article 33(1)) or to a consumer (Article 33(2)) relevant safety information, available to you, when both the following conditions are met:
- The substance is included in the Candidate List for authorisation, and
- The substance is present in the article produced and/or imported above a concentration of 0.1% (w/w).
The information is to be provided to the recipient of the article, i.e. industrial or professional users and distributors, when the article is supplied for the first time after the inclusion of the substance into the Candidate List and to the consumer upon request by that consumer, within 45 calendar days of that request and free of charge.
The communication of information at the request of a consumer is independent of whether the article was purchased by that particular consumer.
The information you should communicate must be sufficient to allow safe use of the article, considering all life cycle stages of the article including disposal. Section 3.4.1 of the Guidance on requirements for substances in articles describes in detail which information should be communicated.
If no particular information is necessary to allow safe use of the article containing a Candidate List substance, e.g. when exposure can be excluded at all life cycle stages of the article including disposal, as a minimum the name of the substance in question has to be communicated to the recipients of the article or to consumers.
The Guidance on requirements for substances in articles is available on the ECHA website at: http://echa.europa.eu/guidance-documents/guidance-on-reach
In the Guidance on requirements for substances in articles, the term “complex object” refers to any object made up of more than one article. In complex objects, several articles can be joined or assembled together in various manners. For example, they can be mechanically assembled (e.g. pair of (metallic) scissors, foldback clips) or joined using substance(s)/mixture(s) (e,g. block of sticky notes, glued chip in a bank card, unpainted bicycle frame formed by welding together multiple steel tubes). The more articles it is made of, the more complex an object becomes.
Articles that are assembled or joined together in a complex object remain articles, as long as they keep a special shape, surface or design, which is more decisive for their function than their chemical composition, or as long as they do not become waste (as defined in the Waste Framework Directive - Directive 2008/98). An importer or a supplier of a complex object (e.g. foldback clip) is importer or supplier of the various articles the complex object is made from, and must therefore comply with the applicable communication and notification obligations for each one of them (e.g. the bent strip of steel and the two metallic wire handles of the foldback clip). Whether the complex object is an article depends on whether it fulfils the criteria of the definition of an article.
The determination of the concentration of a Candidate List substance, to check whether it is above the 0.1% w/w threshold or not, is essential to check whether communication and notification obligations apply.
Table 5 in the Guidance on requirements for substances in articles illustrates and explains in detail several scenarios on how to determine the concentration of a Candidate List substance (weight by weight (w/w)) in an article or in articles incorporated in a complex object (any object made up of more than one article). In summary:
- Scenario I. Article made from a Candidate List substance as such or in a mixture: the concentration is calculated over the total weight of the article (e.g. plastic chair produced by injection moulding)
- Scenario II. Candidate List substance as such or in a mixture used for joining two or more articles (complex object): the concentration of the Candidate List substance is calculated over the total weight of the complex object (e.g. unpainted bicycle frame formed by welding together multiple steel tubes, using a mixture containing the substance)
- Scenario III. Candidate List substance in coatings (e.g. paint, lacquer, varnish, functional coating) - III. A) Fully coated article; III. B) Partially coated article; III. C) Coated complex object: the concentration of the Candidate List substance in the (fully/partially) coated article is calculated over the total weight of the coated article or of the complex object
- Scenario IV. Very complex objects (e.g. sofa, bicycle, mobile phone, car and aircraft): the calculation rules set out for scenarios I to III above apply for each article or simpler complex object.
The Guidance on requirements for substances in articles is available on the ECHA website at: http://echa.europa.eu/guidance-documents/guidance-on-reach
The obligations of the importer of the mixture depend on the interpretation of each specific entry in Annex XVII to REACH for the substance concerned, taking account of the wording, the context and the purpose of the restriction in question.
For instance, if a substance were completely banned, then it could not be placed on the market, not even as an impurity in a substance in an imported mixture. On the other hand, some Annex XVII entries specify limits above which a substance cannot be placed on the market. This limit may not be exceeded, no matter what is the source of the substance in the mixture. However, this can only be determined on a case-by-case basis depending on the substance, the restriction and the concentration of the substance as an impurity in the imported mixture.
It should be noted that the impurity may be permissible at any concentration if the use of the imported mixture is not covered in the ‘conditions of restriction' listed in Annex XVII for the substance.
In special cases where, due to the size or shape of the packaging, it is technically not possible to include the protective gloves inside the packaging, it is considered to be sufficient that the gloves are fixed tightly to the packaging in a manner that they cannot be unintentionally removed during handling and transport. The gloves must not obstruct the label and the removal of the gloves must not destroy the label. In addition, both the packaging containing the mixture and the protective gloves must be placed on the market as a single unit, which explicitly signals to the consumer that the mixture may only be used with the protective gloves .
The new legal act may explicitly state that references to the repealed act must be construed as references to the new legal act (e.g., Article 139 of REACH).
For example:
- Entry 19 (paragraph 4) exempts certain uses of arsenic compounds for wood preservation if they are authorised in accordance with Directive 98/8/EC. This Directive was replaced by Regulation (EU) 528/2012, which explicitly states that references to the repealed Directive must be interpreted as reference to the new regulation (Article 96).
- Entry 45 (paragraph 3) exempts electrical and electronic equipment within the scope of Directive 2002/95/EC from the restriction of diphenylether, octabromo, derivative. This Directive was replaced by Directive 2011/65/EU, which explicitly states that references to the repealed Directive must be interpreted as reference to the new directive (Article 26).
- Entry 50 (paragraph 3) defines tyres covered by the PAH restriction as tyres for vehicles covered by three directives, including Directive 2002/24/EC. This Directive was replaced by Regulation (EU) 168/2013, which expressly states that references to the repealed Directive must be interpreted as reference to the new regulation (Article 81).
Article 31(6) of the REACH Regulation provides that the safety data sheet (SDS) shall contain a Section 15 entitled ‘regulatory information’. Annex II to REACH provides requirements for the compilation of the SDS. Section 15(1) specifically mentions that, if the substance or mixture covered by the SDS is the subject of specific provisions in relation to the protection of human health or the environment at Union level (e.g. restrictions under Title VIII), these provisions must be mentioned, unless this information is already mentioned in other parts of the SDS. Thus, all restriction entries applicable to the specific substance or mixture covered by the SDS need to be indicated therein. As an example, SDSs including carcinogenic, mutagenic or toxic to reproduction substances (as such or in a mixture) listed in appendices 1 to 6 to REACH, need to refer to entries 28, 29 or 30 of Annex XVII. If another specific restriction exists for these substances, this needs to be mentioned as well in the SDS.
Moreover, Article 31(9) of the REACH Regulation requires suppliers to update the SDS without delay once a restriction has been imposed. In Section 16 (other information), a clear indication of where changes to the previous version have been made needs to be included, unless such indication is given elsewhere in the safety data sheet, with an explanation of the changes.
To further clarify the exemption (within Article 67(1) of the REACH Regulation, manufacture, placing on the market or use of a substance in scientific research and development (SRD) is exempted for restrictions), note that under the authorisation process the following Q&As (concerning the exemption in Article 56 for the use of Annex XIV substances in scientific research and development) has been provided. The same approach can be broadly considered as applicable to restrictions.
- Q&A 1153 states that sampling for further analysis is not exempted and thus not regarded as scientific research and development. However, “activities considered to form part of the use of the sample in performing analytical activities” fall within the exemption.
- Q&A 1030 explains that the uses of a substance upstream preceding an exempted end-use in scientific research and development are also exempted in quantities of the substance ending up in SRD (i.e. under 1 t/y per user) subject to certain conditions.
- Q&A 585 explains that the exemption from authorisation also applies to the use of a substance in analytical activities such as monitoring and quality control under certain conditions. This exemption applies irrespective of where the analysis is performed i.e. on-site or off-site facilities, but does not cover sampling activities.
Many entries in the Restriction List (Annex XVII) cover articles. Such entries are, for instance, entries 50 - 52, 61 and 63. These may address types of textiles and leather articles, even if these are not explicitly mentioned.
The following entries in the Restriction List (Annex XVII) are specific to textiles:
- Entry 4 ((2,3 dibromopropyl) phosphate, CAS No 126-72-7);
- Entry 7 (Tris(aziridinyl)phosphinoxide, CAS No 545-55-1; EC No 208-892-5) and
- Entry 8 (Polybromobiphenyls; Polybrominated biphenyls (PBB), CAS No 59536-65-1).
These entries state that these substances “Shall not be used in textile articles, such as garments, undergarments and linen, intended to come into contact with the skin.”
The following entries in the Restriction List (Annex XVII) restrict substances in relation to textiles and/or leather articles:
- Entry 18, restriction on mercury compounds in the impregnation of heavy-duty industrial textiles and yarn intended for their manufacture;
- Entry 20 (paragraph 6), restriction on dioctyltin (DOT) compounds in textile articles intended to come into contact with the skin;
- Entry 23 (paragraph 6), restriction on cadmium and its compounds in textiles and clothing;
- Entry 43, restriction on azocolourants and azodyes in textile and leather articles which may come into direct and prolonged contact with the human skin or oral cavity (indicative list is provided);
- Entry 46 (paragraph 3) restriction on nonylphenol and nonylphenol ethoxylates in textiles and leather processing (with some exceptions);
- Entry 46a, restriction on nonylphenol ethoxylates in textile articles which can reasonably be expected to be washed in water during their normal lifecycle, and
- Entry 47 (paragraphs 5-7), restriction on chromium VI compounds in leather articles coming into contact with the skin; and
- Entry 72 (paragraph 1), restriction on specific substances listed in column 1 of the Table in Appendix 12 in i) clothing and related accessories, ii) textiles other than clothing which, under normal or reasonably foreseeable conditions of use, come into contact with human skin to an extent similar to clothing and iii) footwear if they are for use by consumers (with some exceptions).
The following entries of the Restriction List (Annex XVII) specifically include a derogation for electrical and electronic equipment:
Entry 45 (paragraph 3) provides a derogation from the restriction on diphenylether, octabromo derivative (C12H2Br8O) for electrical and electronic equipment within the scope of Directive 2002/95/EC, which has been replaced by Directive 2011/65/EU (on the restriction of the use of certain hazardous substances in electrical and electronic equipment), and
Entry 63 (paragraph 8) excludes articles within the scope of Directive 2011/65/EU from the restriction on lead and its compounds in articles supplied to the general public.
In addition, the following entries in the Restriction List (Annex XVII) explicitly restrict the placing on the market of paints and/or paint strippers:
Entry 16, restriction on certain lead carbonates in substances or mixtures intended for use as paint;
Entry 17, restriction on certain lead sulphates in substances or mixtures intended for use as paint;
Entry 20, restriction of organostannic compounds acting as biocide in free association paint;
Entry 20 (paragraph 5), restriction on dibutyltin (DBT) compounds in paints and coatings;
Entry 23 (paragraph 2), restriction on cadmium and its compounds in paints with codes [3208] and [3209] and in painted articles;
Entries 28-30 (paragraph 1), restriction on CMRs as substances, as constituents of other substances or in mixtures (including paints), for supply to the general public; (paragraph 2) derogation for artists’ paints covered by Regulation (EC) No 1272/2008;
Entry 48, restriction on toluene (CAS No 108-88-3; EC No 203-625-9) in spray paints intended for supply to the general public;
Entry 54, restriction on (2-(2-methoxyethoxy)ethanol (DEGME) (CAS No 111-77-3; EC No 203-906-6) for supply to the general public, as a constituent of paints and paint strippers;
Entry 55, restriction on 2-(2-butoxyethoxy)ethanol (DEGBE) (CAS No 112-34-5; EC No 203-961-6) for supply to the general public, as a constituent of paints and paint strippers and in spray paints and
Entry 59, restriction on dichloromethane in paint strippers under certain conditions.
In general, cleaning refers to any removal of dirt or pollution from articles or places, both in industrial and institutional facilities as well as in households.
In some entries cleaning may be specified by reference to a particular user group(s) or potential exposure pattern to which the restriction applies. For example, column 2, paragraph 1 of entries 32-38 refer to the substance or mixture being intended for supply to the general public and/or intended for diffusive applications where releases can occur from multiple sources such as surface cleaning and cleaning of fabrics. Column 2, paragraph 1 of entry 46 applies to industrial and institutional cleaning systems (e.g. in schools, hospitals), with the exception of dry cleaning done in a controlled closed system where the washing liquid is recycled or incinerated and other cleaning systems where special treatment is used with recycling or incineration of the washing liquid. Column 2, paragraph 2 entry 46 applies to all domestic cleaning.
The term ‘article’, as interpreted by the European Court of Justice (ECJ) in its judgement of 10 September 2015 in case C-106/14 applies in the same way to restrictions in Annex XVII as to the other aspects of the REACH Regulation. The judgement is available here: http://curia.europa.eu/juris/liste.jsf?language=en&td=ALL&num=C-106/14.
For the purposes of REACH, the term 'article' has the specific meaning set out in Article 3(3) of the REACH Regulation. Article 3(3) defines an article as ‘an object which during production is given a special shape, surface or design which determines its function to a greater degree than does its chemical composition’.
'Complex objects' are made up of more than one article which meet the criteria laid down in Article 3(3) of REACH, e.g. a bicycle is a complex object made up of several articles, such as handlebar grips, cables, screws etc. Complex objects are explained in the ECHA Guidance on requirements for substances in articles. Please note the term ‘complex object’ corresponds to ‘complex product’ that is used in the ECJ Judgement referred to above (see footnote 12 of the above guidance).
The ECJ, in its judgment, observed that the REACH Regulation does not contain any provisions specifically governing complex products and that consequently, in the absence of specific provisions, there is no need to draw a distinction between articles of their own (e.g. screw) or when incorporated as components of a complex product (e.g. a screw in a bike). Therefore, when incorporated into a complex product, an 'article' remains an article within the meaning of REACH, as long as the article retains its special shape, surface or design, which is more decisive for its function than its chemical composition.
Entries in Annex XVII restricting 'articles' cover any object meeting the criteria in Article 3(3) of the REACH Regulation. Therefore, if an entry restricts the placing on the market of articles containing/releasing substance X and the restriction affects complex objects, the presence/release of substance X in each of the individual articles should be checked.
If a restriction entry refers to ‘parts of articles’, this should be understood as referring to an integral part of an article. Please note questions and answers (Q&As) and guidelines for some entries in Annex XVII have been developed which provide further information. The Q&As and guidelines are available on the ECHA website here: https://echa.europa.eu/information-restricted-substances.
A number of Annex XVII entries (Entries 3, 20, 28-30, 32-38, 40, 48, 50, 54, 55, 56, 57, 59, 63(7) and 73) specifically refer to the ‘supply to the general public’ in their conditions to the restriction. Substances, mixtures or articles are considered to be for supply/supplied to the general public if they are to be placed on the market for end-users belonging to that group (e.g. final consumers).
The fact that a substance, mixture or article is supplied between economic operators before arriving to the general public, does not exempt it from any of the conditions (e.g. concentration limits) imposed by Annex XVII entries that make reference to ‘for supply to the general public’. Also substances, mixtures or articles put at the disposal of the general public in a public area fall within the definition of ‘supply to the general public’, irrespective of the type of ownership (public or private) or transaction by which they were put at their disposal. As an example, in the restriction entry 50, paragraph 5, it is understood that tiles/mats used in public playgrounds are supplied to the general public when they are put at the disposal of the general public.
It is to be noted that some restriction entries have general restriction ‘placing on the market’ without referring to any specific group, thus covering also supply to the general public (e.g. entry 63(1)). Entry 69 refers to ‘placed on the market to the general public’, which is considered to have similar meaning than ‘supply to the general public’.
Substances identified as Persistent Organic Pollutants (POPs) are regulated worldwide by the Stockholm Convention and the Aarhus Protocol. These international agreements are implemented in the EU by Regulation (EU) No 2019/2021 (POPs Regulation).
When a POP substance is included in the Annexes I, II, III or IV to the Regulation (EU) 2019/2021, the POPs Regulation applies. The manufacturing, placing on the market and use of substances listed in Annex I or II of POPs is prohibited or severely restricted (some exemptions apply).
When a substance already restricted under REACH is included in Annex I or II to POPs, the Commission removes the corresponding entry from Annex XVII of REACH by a separate amendment to REACH. For example, substances that were included in restriction entries 22, 42, 44, 53 and 67 of Annex XVII to REACH have been identified as POPs and these entries have been deleted. PFOA substances included in restriction entry 68 have also been identified as POPs and restriction entry 68 has been amended by removing PFOAs and including C9-14 PFCAs.
You can find information on the deleted entries and the related Commission Regulations under ‘Further information on the table’ on the ECHA website in the Substances restricted under REACH section.
Substances included into an Annex to the POPs Regulation should be removed from Annex XVII of REACH. However, it may happen that the inclusion into the POPs list and deletion from Annex XVII do not occur simultaneously. In such a case, the most restrictive conditions (usually under the POPs Regulation) apply.
Restriction entry 68 was first included into Annex XVII to REACH by Commission Regulation (EU) 2017/1000 on 14 June 2017 and it initially covered Perfluorooctanoic acid (PFOA), its salts and PFOA-related substances. PFOA substances have been further included in the list of substances restricted under the POPs Regulation (Regulation (EU) 2019/1021) in June 2020 and consequently removed from Annex XVII of REACH. A new entry 68 of Annex XVII to REACH (same entry number but new substances) was subsequently included by Commission Regulation (EU) 2021/1297 of 4 August 2021, as corrected, which restricts the placing on the market and use of perfluorocarboxylic acids containing 9 to 14 carbon atoms in the chain (C9-C14 PFCAs), their salts and C9-C14 PFCA-related substances.
A plastic article is an article made entirely of plastic materials or an article containing components constituted by plastic materials. REACH Regulation doesn’t define plastic, however a definition of plastic is available in Article 3(1) of Directive (EU) 2019/904 as: ‘a material consisting of a polymer as defined in point 5 of Article 3 of Regulation (EC) No 1907/2006, to which additives or other substances may have been added, and which can function as a main structural component of final products, with the exception of natural polymers that have not been chemically modified’.
In the REACH regulation, a number of restriction entries apply directly to articles made of plastic materials. There are other entries that apply to articles in general, that include also articles made of plastics. However, the possible presence of certain restricted substances in plastic articles has to be assessed on a case-by-case basis.
Entries related mostly to plastic articles:


Entries related to specific categories of articles:


Restriction Entries covering all articles:


ECHA has developed a ‘Compendium of Analytical Methods’ to provide a ready reference of some available analytical methods that authorities or industry may use in order to assess the compliance of chemicals manufactured, used or placed on the Union market with the restrictions set forth in Annex XVII to REACH. If a test method is specified in the legal text, it has to be adhered to.
For instance, for Entry 27 (Nickel and its compounds) there are 3 available analytical methods recommended by the ECHA Forum that can be used in the verification of compliance of this entry:
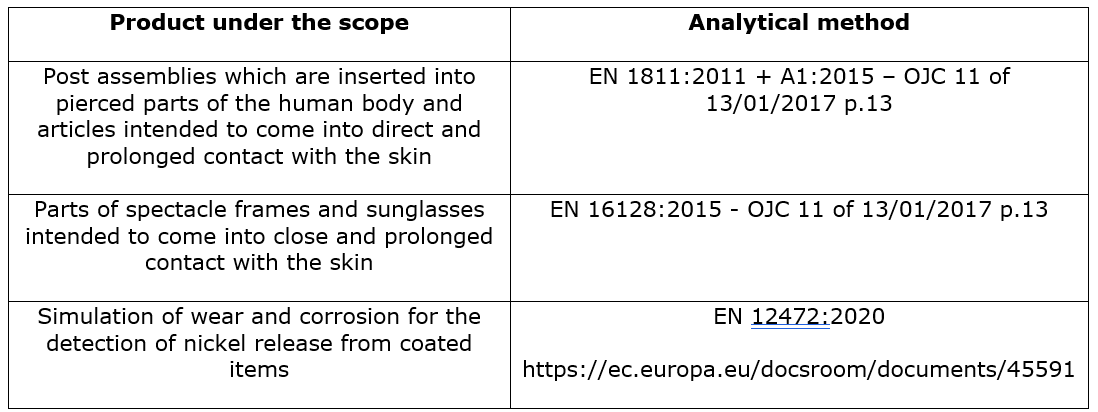
As of April 2021, the titles and references of standards to be used as the analytical methods for demonstrating the conformity of articles under entry 27 of Annex XVII to REACH will be published on the Commission website only and no publication in the Official Journal of the European Union is foreseen anymore. Furthermore, updated references and titles of standards relevant for entry 27 can be found in both the restrictions webpage and the harmonised standards webpage of DG GROW.
Concerning the Restriction on CMRs 1A and 1B in Textiles and Clothing, a list of available analytical is available in the document Explanatory Guide on the Restriction on CMRs 1A and 1B in Textiles and Clothing.
Lastly, information on test methods is available in each of the background documents accessible in the Registry of restriction intentions.
A specific frequency for the update of Annex XVII is not indicated in the REACH regulation. The List of restricted substances (Annex XVII, REACH) is updated by amendments of the REACH Regulation. These amendments may result in the introduction of a new restriction entry, modifications or deletion of an existing entry.
The timelines for amending Annex XVII of REACH are in the remit of the European Commission. In addition, Annex XVII contains certain descriptive entries covering substances listed in appendices, which are also updated when new substances fall under the respective entries (e.g., when new harmonised classifications are laid down for substances and which have been included in the appendices by the Commission Regulation). It is possible to follow the changes of a given entry throughout the years under ‘History’ in the entry-specific web page on the ECHA’s website in the Substances restricted under REACH section.
In addition, ECHA's website includes a Registry of restrictions intentions in order to allow interested parties to be aware of the substances for which the authorities intend to submit Annex XV dossiers for restrictions.
Article 67(1) of the REACH Regulation states that a substance in its on, in a mixture or in an article, for which Annex XVII contains a restriction, cannot be manufactured, placed on the market or used unless it complies with the conditions of that restriction.
On the basis of Article 69(1) of REACH the Commission may request, ECHA, to start a restriction procedure when they are concerned that the manufacture, placing on the market or use of a certain substance poses a risk to human health or the environment that is not adequately controlled and needs to be addressed. A Member State (or a group of Member States) can also start a restriction procedure on the basis of Article 69(4) of REACH.
On the Basis of Article 69(2), when the sunset date of a substance included into Annex XIV or REACH (Authorisation List) has passed, ECHA verifies if the use of that substance in articles may pose a risk to human health or the environment. If ECHA concludes that the risk is not adequately controlled, they have to submit a restriction proposal (Annex XV dossier) to the Commission.
The restriction procedure applicable to these cases is regulated in Articles 69-73 of REACH and explained on the ECHA website in the Restriction procedure page.
Note that to communicate about the upcoming proposal in advance, the intention to prepare a restriction proposal is made public in the registry of intentions before the proposal file itself is prepared.
The phases (and their duration) of this restriction process are:
1) Preparation and submission of a restriction proposal (12 months) – Phase 1
2) Public consultations on the restriction report and the SEAC’s draft opinion (6 months) – Phase 2a
3) Opinion development of ECHA’s Committee for Risk Assessment (RAC) and Committee for Socio-economic Analysis (SEAC) (12 months since publication of the proposal) – Phase 3.1
4) If the Commission considers that the conditions for a restriction are fulfilled, it prepares a draft amendment to the list of restrictions in Annex XVII to REACH (within 3 months of receipt of the SEAC opinion). Commission decision in a comitology procedure (duration varies). And finally, complying with and enforcing the restriction – Phase 3
The Commission can also propose restrictions for consumer use as regards substances on their own, in mixtures or in articles, which meet the criteria for classification as carcinogenic, germ mutagenic or toxic for reproduction category 1A or 1B and could be used by consumers (Art. 68(2) REACH). This process follows a simplified path which does not require consultations by RAC and SEAC.
Interested parties can follow all phases of a restriction proposal from the notification of the intention, to the adoption of the final opinions by the Committee for Risk Assessment (RAC) and the Committee for Socio-economic Analysis (SEAC), till the adoption of the restriction by the European Commission by amending Annex XVII of REACH.
Stakeholders are encouraged to submit any relevant information to the dossier submitters during the preparation of the restriction proposal by responding to specific Call for Evidence published in ECHA’s website or in the website of the Member State Authority who is preparing the proposal.
Once the proposal is submitted, information can be provided to support the decision-making process by responding to the public consultations on the restriction proposal (once it is submitted) and on SEAC draft opinion. Advice on the public consultation and dedicated procedure on how to submit information are provided in the ECHA’s website.
More information on the restriction process can be found on the ECHA website.
This entry refers to hazard classes set out in Regulation (EC) No 1272/2008 on the classification, labelling and packaging of substances and mixtures (CLP Regulation). A full list of the hazard classes referred to in this entry with their corresponding hazard categories and hazard statements is provided in this table.
The REACH Regulation does not provide a specific definition for paints.
According to a general meaning, paint is a mixture, usually of a liquid with a solid pigment. Furthermore, Commission Decision 2009/544/EC (establishing the ecological criteria for the EU Ecolabel to indoor paints and varnishes) provides the following definition: 'Paint' means a pigmented coating material, in liquid or in paste or powder form, which when applied to a substrate, forms an opaque film having protective, decorative or specific technical properties.
The abovementioned definitions provide an indication of what could be generally considered as a paint in the context of entries 16-17 of Annex XVII (to REACH) provisions.
It should be noted that the restrictions in entries 16-17 of Annex XVII only allow Member States to permit the use of paints containing the restricted substances for the restoration and maintenance of works of art and historic buildings and their interiors. Thus children's paint sets and other stationery-type paints such as artist paints and do-it-yourself (DIY) decorations for t-shirts must not contain the restricted substances.
Concerning children's paint sets, these may also be covered by the Toys Directive (Directive 2009/48/EC on the safety of Toys), which lays down limits for the presence of lead in toys.
The REACH Regulation does not provide a definition of measuring devices (often referred to as measuring instruments). However, in the context of entry 18(a), it has to be noted that:
- The restriction provision in paragraph 1 of entry 18(a) covers those measuring devices that are intended for sale to the general public and provides an indicative list of these (thermometers, manometers, barometers, sphygmomanometers etc.)
- Furthermore paragraphs 5 and 7 provide exhaustive lists of mercury-containing measuring devices intended for industrial and professional uses which have not been allowed to be placed on the market after 10 April 2014 (barometers; hygrometers; manometers; sphygmomanometers; strain gauges to be used with plethysmographs; tensiometers; thermometers and other non-electrical thermometric application; mercury pycnometers; mercury metering devices for determination of the softening point; mercury triple point cells other than those used for the calibration of platinum resistance thermometers).
For the purposes of Directive 2004/22/EC on measuring instruments, ‘measuring instrument' means any device or system with a measurement function that is covered by Articles 1 and 3. According to the European Standardisation organisation-CEN, "measuring instruments allow for testing the accuracy and calibration of measuring devices (e.g. water meters, gas meters, electricity meters, etc.)". (http://www.cencenelec.eu/standards/sectors/mid/pages/default.aspx)
The abovementioned definitions provide an indication of what could generally be considered as a measuring device in the context of entry 18(a) of Annex XVII to REACH provisions.
Paragraph 4b) of Annex XVII to REACH concerning arsenic compounds provides for a list of applications for which wood treated with CCA may be used. This is not a list of examples of possible uses but an exhaustive list of authorised applications. It flows both from the actual wording of those provisions and from their objective that the derogation provided for in paragraph 4 must necessarily be subject to a strict interpretation, as confirmed by the Court of Justice (case C-358/11, pp. 40-43).
Consequently, wood treated with CCA cannot be used for other applications than the ones listed in paragraph 4 b). Wood treated with CCA can, therefore, not be used for railway sleepers installed above ground.
Organostannic compounds covered by entry 20 in Annex XVII to REACH, must contain a carbon-tin bond. Substances like tin salts or organotin compounds, for which tin is bound to an atom other than carbon (for example hexanoic acid, 2-ethyl-, tin(2+) salt (CAS-No. 301-10-0)) are not covered by entry 20 in Annex XVII to REACH.
(a) Yes, they do. Entry 20 in the Restriction List (Annex XVII) to REACH imposes DOT compounds restrictions for childcare articles, which are not toys. The REACH Regulation does not contain a definition of toys. The definition of toys in Directive 2009/48/EC on the Safety of Toys is illustrative in determining what should be considered as a “toy” in the context of this restriction (see Q&A 0982).
However, paragraph 6(a) of entry 20 restricts DOT compounds in textile toys as in any other textile article intended to come into contact with the skin. Furthermore, organic tin (including DOT) in toys is restricted by paragraph 13 of Part III (Chemical Properties) of Annex II to Directive 2009/48/EC on the safety of toys, which specifies maximum migration limits.
(b) and (c)
In addition to derogation in paragraph 5(d), the other derogations currently applicable to TBT, DBT and DOT compounds in articles relate to the continued placing on the market of articles that were already in use in the EU before the 1 July 2010 for TBT (paragraph 4(b), before the 1 January 2012 (paragraph 5(b)) for DBT and before the 1 January 2012 for DOT (paragraph 6(b)).
Yes. Packaging can be considered as an article (or a product composed of different articles) in its own right. More information can be found in the ECHA guidance on requirements for substances in articles available at https://echa.europa.eu/guidance-documents/guidance-on-reach (in particular in chapter 2).
Therefore, with regard to entry 20, packaging should comply with the restrictions for tri-substituted (TBT, TPT) and dibutyltin (DBT) compounds. The restriction of dioctyltin compounds (DOT) which applies only to certain listed articles for the general public, applies to textile packaging.
Compliance with the restriction is assessed based on the weight of the entire painted article and not based on the weight of the layer of dry paint. This approach follows the scenarios from the Guidance on requirements for substances in articles, Table 5 under subchapter 3.2.3.1.
For further details see Q&A 1564 on Annex XVII and concepts of ‘article’, ‘complex object’ and ‘parts of articles’.
With reference to paragraph 10 of the Annex to Commission Regulation (EU) 494/2011 amending entry 23 of Annex XVII of the REACH Regulation (cadmium) the concentration threshold of cadmium applies in each metal part of jewellery. The wording used by the legislator, i.e. "metal parts of the jewellery and imitation jewellery" implies that each metal part is relevant; therefore in order to determine if the restriction applies the calculation of the concentration in this case is to be done for each metal part. Therefore, if there are several metal layers as coatings on the surface of an inner (metallic) part of the jewellery these should be regarded as integral part of the metal part and the concentration limit of 0,01% is calculated for this whole metal part. In case the inner part is not metal, but the coating is made of metal layers, this coating is regarded as one metal part. If the jewellery article contains several metal parts, each of them should comply with the concentration limit.
The prohibition of the placing on the market of jewellery and imitation jewellery articles containing cadmium includes sales from the manufacturers to distributors and from distributors to retailers, as well as imports. However, Commission Regulation (EU) 494/2011 contains derogation for articles that were placed on the market before 10 December 2011 (for the date see corrigendum published in OJ L 136/105). This means that jewellery and imitation jewellery articles placed on the market for the first time before 10 December 2011 do not need to comply with the prohibition thus they can be sold following entry into force of the new restriction for example to a retailer or on the second-hand market.
Paragraph 8 of entry 23 of Annex XVII to the REACH Regulation states that cadmium and its compounds shall not be used in brazing fillers in a concentration equal to or greater than 0,01 % by weight. In addition, brazing fillers shall not be placed on the market if the concentration of cadmium (expressed as Cd metal) is equal to or greater than 0,01 % by weight. The paragraph also states that brazing shall mean a joining technique using alloys and undertaken at a temperature above 450°C. In accordance with the following paragraph 9, by way of derogation paragraph 8 shall not apply to brazing fillers used in defence and aerospace applications nor to brazing fillers used for safety reasons.
The safety aspect in relation to this derogation is if the use of cadmium containing brazing filler may prevent accidents causing human suffering or environmental pollution.
For the enforcement purposes examples of applications are given. It can be considered that the derogation on uses of cadmium containing brazing fillers for safety reasons in paragraph 9 of entry 23 covers the current uses, such as:
-
Brazing fillers used in turbine wheels in power plant technology in temperature below 650°C.
Turbine wheels in power plant technology are parts of speed drivers for gas compressors and boiler feed pumps, where rotational speed is approximately from 1000 revolutions per minute (rpm) up to 20 000 rpm. Cadmium containing brazing fillers are needed as they can be used below 650 °C without decreasing the strength of the parent material (base metal). Cadmium-free brazing fillers require higher temperatures which causes the weakening of the parent material. Weakening of the parent material could lead to the breakdown of the turbine wheel. Due to high rotational speed the parts and pieces of shrapnel may cause injuries to workers and others in the vicinity of the wheel. The breakdown of the turbine wheel may result also in a complete shutdown of the power plant, the compressor station of a gas pipeline, or of a refinery.
-
Brazing fillers used in pipes and tubes where acetylene is transferred in high pressure (1.5 – 17 bar).
Cadmium containing brazing fillers are needed in the joining process for pipes and tubes, where acetylene is transferred in order to avoid formation of explosive substances. Acetylene forms explosive substances with copper and silver as well as other materials (e.g. formation of copper acetylide and silver acetylide). Cadmium reduces the overall percentage of copper and silver in the brazing fillers to the level where formation of explosive substances does not exist. Another reason to use cadmium in brazing fillers for this application is that cadmium facilitates the capillary action and solder penetration ensuring a good quality joint with high integrity for pipes and tubes where acetylene is transferred in high pressure (1.5-17 bar). Release of acetylene from pipes and tubes may as well cause serious risk, as acetylene is extremely flammable gas and explosive with and without contact with air.
Other applications that would like to benefit from the derogation need to show the similar kind of safety aspects as described above.
Such considerations should take into account the availability on the market of cadmium-free brazing fillers which can address the safety aspects of the specific application of the brazing fillers in an equivalent manner.
See also ECHA´s report "The use of brazing fillers containing cadmium for safety reasons" [PDF].
Paragraph 1 of entry 23 of Annex XVII to the REACH Regulation provides a restriction on cadmium and its compounds in mixtures and articles produced from certain synthetic organic materials (plastic materials) and paragraph 2 provides a restriction on cadmium and its compounds in paints (Tariff codes 3208 and 3209). The following paragraph 3 states that by way of derogation the restrictions in paragraphs 1 and 2 do not apply to articles coloured with mixtures containing cadmium for safety reasons.
There are two safety aspects in relation to this derogation. The first relates to the use of a specific colour or pigment with certain properties which is necessary to prevent accidents. The second relates to the use of a specific colour or pigment with certain properties in safety equipment.
For the enforcement purposes example of applications are given. Based on above, it can be considered that the derogation in paragraph 3 of entry 23 covers current applications of articles such as:
-
Coloured wire insulation and cable jackets used in aircraft electrical and control systems for the purpose of fire detection and extinguishing systems, flight control systems or during flight tests.
The wire and cable connections are often used in in a high temperature application (greater than 150ºC ambient temperature). Cadmium pigments are used to keep the colour from changing or fading over time in the high temperature. Changing established colour conventions could introduce a significant risk of maintenance errors, which may lead to a risk of passengers.
-
Outdoor safety equipment, such as:
- parts of rescue boats for ships (e.g. safety belts, water pockets of life rafts, canopies) and
- parts of safety equipment for outdoor applications (e.g. seats, reels and diverse technical parts).
The other applications that would like to benefit from the derogation need to show the similar kind of safety aspects than described above.
Such considerations should take into account the availability on the market of alternative substances which can address the safety aspects of the specific application in an equivalent manner.
See also ECHA´s report "The use of cadmium and its compounds in articles coloured for safety reasons" [PDF].
Entry 23 (paragraph 10 (i)) of Annex XVII to the REACH Regulation states that cadmium and its compounds must not be used or placed on the market if the concentration of cadmium is equal to or greater than 0,01 % by weight of the metal in metal beads and other metal components for jewellery making (see Q&A, 158). In addition, it should be noted that articles produced from plastic material referred to in paragraph 1 of the entry must not be placed on the market if the concentration of cadmium is equal to or greater than 0.01 % by weight of the plastic material.
Thus, in the case of plastic coated metal beads (CCB beads), both the plastic material and the metallic part of the bead need to comply with entry 23 (cadmium restriction).
Please also note that if the article is painted, then paragraph 2 will also apply to this article.
Note that metallic articles containing cadmium may be covered under other EU legislation. For example, (1) electrical and electronic equipment falls under Directive 2011/65/EU on the Restriction of the use of certain Hazardous Substances and must comply as well with the maximum concentration limits for cadmium set in that Directive and (2) toys are covered under Directive 2009/48/EC on the Safety of Toys.
Entry 27 of Annex XVII to REACH states that nickel may not be used "in articles intended to come into direct and prolonged contact with the skin, if the rate of nickel release from the parts of these articles coming into direct and prolonged contact with the skin is greater than 0.5 Jg/cm²/week". The aim of this restriction to protect consumers against nickel allergy which may be caused by prolonged contact of the skin with nickel-releasing articles that come into direct and prolonged contact with the skin such as jewellery, buttons, tighteners, zips and rivets in items of clothing. It has emerged that some mobile telephones contain nickel in surface material and that consumers are at risk of developing eczema through skin contact with the mobile telephone. As mobile telephones are clearly intended to come into direct contact with the skin, and as they are used on a daily basis often for prolonged periods of time, it is considered that mobile telephones fulfil the condition of "direct and prolonged contact with the skin". Therefore mobile telephones are covered by the restriction and should comply with the conditions set in Entry 27 of Annex XVII to REACH.
Prolonged contact with the skin is defined as contact with the skin to articles containing nickel of potentially more than
- 10 minutes on three or more occasions within two weeks, or
- 30 minutes on one or more occasions within two weeks.
The skin contact time of 10 minutes applies when there are three or more occasions of skin contacts within a two-week time period. The skin contact time of 30 minutes applies when there is at least one occasion within a two-week time period.
This entry covers:
- post assemblies that are inserted into pierced parts of the human body and release nickel at a rate equal or greater than 0,2 μg/cm2/week;
- any article intended to come into direct and prolonged contact with the skin that releases nickel at a rate greater than 0,5 μg/cm2/week from its parts coming into direct and prolonged contact with the skin. The restriction applies regardless of the materials from which the articles are produced.
The term ‘Prolonged contact with the skin’ is clarified in the Q&A 935.
Substances within the scope of entries 28-30 are not allowed to be placed on the market or used for supply to the general public as substances, as constituents of other substances or in mixtures when the concentration is equal to or greater than the specified limits. Certain derogations apply to this restriction as listed in paragraph 2. CMR substances that are present in articles are not within the scope of the restriction imposed by entries 28-30, but other restrictions may be applicable to these substances, when present in articles. In addition, notification and communication obligations under REACH may apply; see the ECHA website: https://echa.europa.eu/regulations/reach/candidate-list-substances-in-articles/notification-of-substances-in-articles.
It should be noted that entries 28 and 29 are applicable to the substances that are listed in Appendices 1 & 2 (carcinogenic (C), categories 1A and 1B) and Appendices 3 & 4 (mutagenic (M), categories 1A and 1B). Entry 30 is applicable to substances which are classified as reproductive toxicants (R), categories 1A and 1B and listed in Appendices 5 and 6. These Appendices are regularly updated by including new substances, after the adoption of a harmonised classification for a substance as CMR, category 1A or 1B according to Regulation (EC) No 1272/2008.
The REACH Regulation does not define aerosols or aerosol dispensers.
According to the ordinary meaning of the word, an aerosol is considered to be "a substance enclosed under pressure and released as a fine spray by means of a propellant gas". (Oxford advanced dictionary definition). The term may in certain contexts be used for a mixture enclosed under pressure and released as a fine spray by means of a propellant gas, or the dispenser or package used to change the ingredient inside the container into a spray by the use of a propellant gas. The European Aerosol Federation uses the term "aerosol" for both the suspension and the dispenser/package.
Furthermore, it should be noted that according to Article 2.3.1 of the CLP Regulation (for "Classification, Labelling and Packaging"), the term "aerosol dispenser" means: any non-reusable container made of metal, glass or plastic and containing a gas compressed, liquefied or dissolved under pressure, with or without a liquid, paste or powder, and fitted with a release device allowing the contents to be ejected as solid or liquid particles in suspension in a gas, as a foam, paste or powder or in a liquid state.
The CLP definition is very similar to the definition provided in Article 2 of the Aerosol Dispensers Directive (ADD) (75/324/EEC).
The above definitions provide an indication of what could be generally considered as ‘aerosols/aerosol dispensers' in the context of Entry 40 of Annex XVII. Note that aerosol generators should be regarded as aerosol dispensers, as the original restriction discusses aerosol generators.
Entry 40 prohibits the use of flammable, highly flammable or extremely flammable substances in "aerosol dispensers where these aerosol dispensers are intended for supply to the general public for entertainment and decorative purposes". Paragraph 1 provides an indicative list of examples of products that are covered by the restriction. These examples are all products to be used to decorate, for instance, venues for festivities (e.g. Christmas, weddings, and carnivals) or parties (e.g. birthday parties and fancy-dress parties) and for entertainment use, for instance, during festivities and parties. None of the examples listed in the entry are cosmetic products within the meaning of Regulation (EC) No 1223/2009 on cosmetic products.
Coloured hair sprays and body glitter would fall within the definition of cosmetic products in Regulation (EC) No 1223/2009, as they are intended "to be placed in contact with an external part of the human body" with a view to "changing its appearance" and therefore have a similar use to more classical cosmetic products, such as normal hair sprays. Coloured hair sprays and body glitter should not be considered as having an entertainment or decorative purpose, and therefore they are not covered by entry 40 of Annex XVII to REACH.
Through a literature search and consultation with experts in this area it was not found any structural connection between optical brighteners (or better called fluorescent dyes) and azodyes since either the NH bonds in the fluorescent dyes are connected to heterocyclic NC structures and therefore cannot form any of the 22 banned arylamines or they do not contain any azo bonds where reductive cleavage could take place to generate any of the aromatic amines covered by the azodyes ban. Therefore at the present time, this information confirms that the restriction in Entry 43 to Annex XVII does not cover optical brightening agents (OBAs). Should the chemical structure of optical brightening agents be different from the definition as reported above, this answer may change accordingly.
Entry 43(3) of Annex XVII restricts the placing on the market of substances and mixtures containing over 0.1% of the azodyes listed in Appendix 9, when they are intended for colouring textile and leather articles, and also the actual use of the substance or mixture for that purpose. Therefore, the presence of these substances in imported articles is not restricted.
However, pursuant to paragraphs 1 and 2 of the restriction, if an azodye in Appendix 9 releases one or more of the aromatic amines listed in Appendix 8 in a concentration above 30 mg/kg (0,003 % by weight, it cannot be used in textile and leather articles which may come into direct and prolonged contact with human skin or the oral cavity (such as those listed in paragraph 1). Those textile and leather articles cannot be placed on the market unless they comply with that concentration limit.
Yes. The intention of the legislator was to cover all isomers, linear and branched, of nonylphenol and their ethoxylates and therefore all of them are covered by the restriction.
The restriction on nonylphenol and nonylphenol ethoxylates was based on the risks identified in the risk assessment report prepared by the United Kingdom under Council Regulation (EEC) No 793/93 of 23 March 1993 on the evaluation and control of the risks of existing substances. The risk assessment report states that "It is understood that nonylphenol (CAS Number: 25154-52-3) as originally defined by CAS (Chemical Abstract Service) covered all nonylphenols. However, subsequent revisions redefined it to cover only straight chain nonylphenol, other isomers having different CAS numbers. Given the method of manufacture of nonylphenols, very little if any straight chain nonylphenol is produced. That which is produced is only likely to be present at very low levels in commercial mixtures. The commercially produced nonylphenols are predominantly 4-nonylphenol with a varied and undefined degree of branching in the alkyl group. This assessment covers commercially produced material (predominantly 4-nonylphenol, branched). This material will also contain smaller amounts of other isomers and impurities, and falls under the CAS Number 84852-15-3."
Therefore Council Directive 76/769/EEC, as amended by Directive 2003/53/EC, did not specify any CAS or EC numbers for nonylphenol. In the revision of the REACH restriction by Regulation 552/2009/EC, which made several technical changes, the CAS and EC numbers were added. This has been corrected by deletion of the EC and CAS numbers from the entry with the Commission Regulation (EU) 2020/2096.
Paragraphs (1) to (9) of entry 46 list the ‘purposes' to which the restriction applies. Following paragraph (7) "cosmetic products" is paragraph (8) "other personal care products". It therefore seems that cosmetics are to be considered as a subcategory of personal care products.
REACH does not define "cosmetic products". According to Regulation (EC) No 1223/2009 on cosmetic products, "cosmetic product" means "any substance or mixture intended to be placed in contact with the external parts of the human body (epidermis, hair system, nails, lips and external genital organs) or with the teeth and the mucous membranes of the oral cavity with a view exclusively or mainly to cleaning them, perfuming them, changing their appearance protecting them, keeping them in good condition or correcting body odours."
Based on the above, it can be interpreted that "other personal care products" within the meaning of entry 46 include, but are not limited to, any product meeting the conditions indicated in the above definition for substances and mixtures and other products used for personal hygiene, such as toilet papers, some female hygiene products, nappies, cotton pads, etc. This interpretation can be broadly considered as a perception of the average consumer for this product category.
The expression ‘textile articles which can reasonably be expected to be washed in water’ relates to the use of care indications on the label attached to the textile article. Textile articles with care instructions that exclude washing in water and require dry cleaning are, in principle, not covered by the restriction in paragraph 1 of entry 46a.
However, care instructions should not be used to circumvent the restriction; this would for example be the case of a label for dry cleaning where there is no need for dry cleaning. ‘New textile articles produced exclusively from recycled textiles’ means that only recycled textiles materials must have been used to produce new textile articles, otherwise the exemption does not apply. If, for example, virgin textiles materials have been used together with recycled ones, the exemption does not apply.
Entry 48 prohibits the placing on the market for supply to the general public of toluene as a substance or in mixtures, in a concentration equal to or greater than 0.1% by weight, where the substance or the mixture is used in adhesives and spray paints. Adhesive tapes consist of a layer of adhesive coated on a flexible substrate. As the restriction concerns the concentration of toluene in adhesives, the concentration of toluene must be calculated with reference to the amount of adhesive on the tape, and not with reference to the total weight of the adhesive and substrate.
As stated in Recital 8 of the Directive 2005/69/EC (OJ L323, 9.12.2005, p.51), there are at present no harmonized test methods for measuring PAHs in the extender oils, or for measuring PAHs in tyres that contain such oils. Until suitable harmonized methods are available, the only named method that is permitted for measuring the PAH content of extender oils is the IP346 analysis method. This method is permitted providing that certain additional conditions are met. These additional conditions are necessary because the IP346 method does not measure the PAH content directly. In fact IP346 measures the total content of polycyclic aromatic compounds (PCA) rather than the PAH content. The PCAs are a group of substances to which PAHs belong, but in which PAHs are present in only very small amounts. The legal limit for PAHs in extender oils, which is 1 part per million (ppm) of BaP and 10ppm total PAH content, is considered to be met if the total PCA content is <3%. In other words, the PCA content of 3% is taken as a proxy measurement for a PAH content of 10ppm. The proxy measurements will be valid only if the ratio between the PAH and PCA content in the extender oil is known and does not change over time. The additional conditions therefore require an initial calibration of the technique (measurement of the PAH/PCA ratio) and recalibration at intervals of six months, or after "major operational change", in order to ensure that the measurements remain valid over time. The term "major operational change" should therefore be taken to mean any change in materials or processes that could invalidate the results of the proxy measurement. The principle cause of invalid results would be a change in the PAH/PCA ratio in the extender oil. However, it should be remembered that not only is IP346 a proxy method for measuring PAH, but that the quantity that it does measure, namely PCA content, is meaaured in a rather indirect way, namely by a change in the refractive index of a solution, and that PCAs are not the only substances that affect the refractive index of a solution. The potential for obtaining invalid results is therefore quite high and the method should therefore be used with considerable caution. It would therefore be advisable to recalibrate in case of doubt. The provision to control the calibration of the PAH/PCA ratio every six months is to safeguard the validity of the IP346 results against unintentional or unknown changes. This would apply for the case where the manufacturing process and materials used remain the same, and where there is no reason be expect a change in the PAH/PCA ratio. However, it is possible to imagine, for example, that a tyre manufacturer receives a reformulated extender oil from his supplier without being made aware of the change that has been made, and the results from the IP346 could be invalidated as a consequence. A six month recalibration interval was considered sufficient to cover such occurrences. Conclusion: The provision to control the calibration of the PAH/PCA ratio after each "major operational change" is to safeguard the validity of the IP346 results. A major operational change is therefore a deliberate change to materials or processes that might be expected to significantly influence the PAH/PCA ratio, or otherwise affect the validity of the measurement. Examples of such a change would be where the source of supply for the extender oil is changed, or where the method of using the oil is changed. Judgment of whether a particular change is sufficiently important to trigger the need for recalibration will necessarily be made case-by-case and will require expert opinion.
Standard reference tyres are produced and imported solely for the purpose of providing a reference performance for other newly developed tyres. They are not placed on the market to be fitted on vehicles intended for final users. For the purpose of entry 50, tyres are defined as tyres for vehicles covered by Directives 2007/46/EC on motor vehicles and their trailers, Directive 2003/37/EC on agricultural or forestry tractors, and Directive 2002/24/EC on two and three-wheeler motor vehicles. It appears that Reference Tyres are not intended to be used on vehicles covered by the Directives 2007/46/EC, 2003/37/EC and 2002/24/EC. In conclusion these tyres should not be considered as covered by the provisions of the restriction in Entry 50 of Annex XVII.
The threshold of 0.1% is the standard threshold used in Annex XVII. The value of 0.1% has been chosen because it represents a measurable quantity. It is being used to take into account impurities, not to allow the use of certain substances, e.g. phthalates in toys and childcare articles. One should be aware that in order to plasticise a toy or childcare article concentrations of phthalates of more than 10 per cent are needed. Different restrictions are applied to each of the two groups of phthalates. The limit value of 0.1% should therefore be applied for each group of phthalates combined, i.e. the concentration of DEHP, DBP and BBP combined should not be higher than 0.1% and the concentration of DINP, DIDP and DNOP combined should also not be higher than 0.1%. Conclusion: A toy or childcare article would not comply with the Entry 51 or Entry 52 respectively if it contained either more than 0.1% of DEHP, DBP and BBP combined or more than 0.1% of DINP, DIDP and DNOP combined. However, it would be considered compliant if it contained only 0.09% of DEHP, DBP and BBP combined and 0.09% of DINP, DIDP and DNOP combined.
The entries 51 and 52 specify that "Childcare article" means "any product intended to facilitate sleep, relaxation, hygiene, the feeding of children or sucking on the part of children." As these articles are intended to facilitate the hygiene of children they should be considered as "childcare articles" as defined by the entries 51 and 52. In conclusion, articles which are used for the hygienic care of children such as bathtubs, articles for the bath, bathtub mats, hairbrushes, bath thermometers, or nail cutters are therefore covered by the Entries 51 and 52 and use of phthalates and should conform to the prescriptions of the entries.
The definition of childcare articles contained in Annex XVII to REACH is as follows: "Childcare articles" are defined as "any product intended to facilitate sleep, relaxation, hygiene, the feeding of children or sucking on the part of children". Further explanation is provided by the Commission services' guidance document on the interpretation of the concept "which can be placed in the mouth". It gives the following examples: "The main purpose of pyjamas is to dress children when sleeping and not to facilitate sleep. Pyjamas should therefore be regarded as textiles and, like other textiles, do not fall under the scope of the Directive. Sleeping bags are designed to facilitate sleep, and should therefore fall under the Directive." Taking this into account, and also taking into account that the guidance document explicitly contains a description and a photo of a mattress cover, it can be confirmed that mattress protectors are childcare articles as defined in Annex XVII. This means that the three phthalates DEHP, BBP and DBP listed in entry 51 of Annex XVII may not be used in mattress protectors. The other three, DINP, DIDP and DNOP, listed in the entry 52, are only restricted in those articles that can be placed in the mouth by children.
The guidance document contains an example of a mattress cover that is not directly mouthable in normal and foreseeable use conditions. The edges and corner are not accessible for mouthing by the child – by design (the mattress should fit snugly in the cot to avoid entrapment risks), and the mattress is covered with a sheet in normal use and the surface is sufficiently taut (by design – to avoid suffocation risks) to prevent PVC from being mouthed through the sheet. This is based on the observation that inaccessible parts of articles can not be taken into the mouth. Articles or parts of articles should be considered inaccessible if, during proper use or reasonably foreseeable improper use by children, they can not be reached. However, there will be other cases when parts of certain articles can be taken into the mouth under normal and foreseeable conditions, for example when the mattress protector is placed on the sheet or cannot be completely fixed. In conclusion, mattress protectors that can be placed above sheets or that cannot be tightly fixed to the mattress have to comply with the restriction contained in entry 52 of Annex XVII to REACH. Authorities competent for market surveillance should assist manufacturers/importers in making a case-by-case assessment on the basis of the criteria described above and in the guidance document.
The restriction in entry 52 concerns the substance "Di-isodecyl phthalate" (DIDP) which is listed with CAS Numbers 26761-40-0 and 68515-49-1. Di-2-propyl heptyl phthalate (DPHP) is an isomer of decyl phthalate and has the CAS No 53306-54-0. According to the information at the Commission's disposal, DPHP (CAS # 53306-54-0) is different from DIDP and therefore not covered by entry 52 of Annex XVII. In conclusion, the substance is not covered under the entry 52 of Annex XVII. The uses of the substance may be regulated in the future on a Community-wide basis, if it appears from the information which will become available that it causes unacceptable risks to human health or the environment. In addition DPHP is explicitly not promoted by its manufacturers for use in toys, food packaging or medical products.
This guideline aims at providing some criteria and examples to help identify those toys and childcare articles which can be placed in the mouth by children. The guideline lists the main criteria of "size dimension" and "accessible parts" (according to the EN 71-European Standard on the safety of toys) which would therefore facilitate the judgement - on a case by case – whether a toy or childcare article can be placed in the mouth by children. The guideline also provides pictures as examples in order to better indicate which toys and childcare articles or parts of them can be taken into the mouth.
The 0.1% limit was set up in order to eliminate the use of phthalates. Such limit was set up in order to be respected in each bit/piece of plasticised material by itself, regardless of whether there are other pieces or not. Therefore the reference to 0.1%, intended also as the sum up to the 3 phthalates, has to be calculated in each piece of plasticised material of each homogenous material. In the example referred to in the question the calculation should therefore be based on the weight of the plasticised material of the head of the doll and not the entire weight of all plasticised material of the whole toy.
A childcare article is defined by entries 51 (4) and 52(4) as "any product intended to facilitate sleep, relaxation, hygiene, the feeding of children or sucking on the part of children" (see Q&A 983). A baby monitor does not facilitate the sleep or relaxation of the baby, it allows parents to be alerted when the baby is crying or making other noise. Therefore baby monitors fall outside the scope of entries 51-52 of Annex XVII.
The polymeric MDI, with CAS number 9016-87-9, is not included in the definition of the substance with CAS 26447-40-5 and moreover is not classified as dangerous in Annex VI of Regulation (EC) No 1272/2008 (CLP). In the Risk assessment report and Risk reduction strategy performed by the Belgian Rapporteur, two main products were analysed during the exposure assessment and throughout the decision making process: the one-component foams and hot melt adhesives, both containing respectively 10% and 2% of MDI. Other products with MDI content below 0.1% did not pose any risk and therefore were excluded by the final consideration on the risk reduction measures under Directive 76/769/EEC. However, polymeric MDI present in mixtures is covered by the restriction when such mixtures contain more than 0.1% of MDI (as defined in the RAR). Therefore, dimers and polymeric forms of MDI are out of the scope of the current restriction except if they are part of mixtures containing more than 0.1% of MDI.
In entry 58, the terms "including natural or legal persons licensed or authorised in accordance with Council Directive 93/15/EEC" should be read as an example of operators that benefit from the exemption. Therefore the derogation in paragraph 2(a) covers all downstream users and distributors as defined in Article 3(13) and 3(14) of REACH. As a consequence, mixtures containing more than 16% of nitrogen in relation to ammonium nitrate may be placed on the market after 27 June 2010 for supply to downstream users and distributors as defined in REACH. As consumers are not downstream users nor distributors, mixtures containing more than 16% of nitrogen in relation to ammonium nitrate may not be placed on the market for supply to consumers.
If a downstream user uses ammonium nitrate to produce a mixture containing ammonium nitrate below the threshold, it may place this mixture on the market for supply the general public. But downstream users should not use ammonium nitrate in order to produce a mixture containing ammonium nitrate above the threshold for supply to the general public. The restriction is applicable to medical devices as well as to other mixtures containing more than 16% of nitrogen in relation to ammonium nitrate for supply to the general public. For example, instant cold packs which contain more than 16% of nitrogen in relation to ammonium nitrate may not be sold to the general public since 27 June 2010.
The restriction is not applicable to downstream users who use ammonium nitrate for their industrial or professional activities in order to transform it into other substances for different purposes. Therefore, for example the use of ammonium nitrate to produce nitrous oxide for use in the production of pressurised foods or for use as anaesthetics is not restricted.
No. Ink strippers, adhesive removers and degreasing agents are not within the scope of the restriction. The restriction covers paint strippers, including also varnish removers and lacquer removers.
This document echa.europa.eu/documents/10162/17220/lead_guideline_information_en.pdf aims at providing a guideline concerning the interpretation of the scope of the restriction provisions in paragraphs 7 to 10 of entry 63 of Annex XVII to REACH Regulation (EU) No 1907/2006 on lead and its compounds in articles supplied to the general public. It has been drawn up to (i) clarify certain terms that define the scope of the restriction (e.g. "accessible part of articles", "normal/reasonably foreseeable conditions of use") (ii) provide non-exhaustive lists of article types (and examples of sub-types) which fall within (or out of ) the scope of the restriction.
Entry 68 applies to
- Linear and branched perfluorocarboxylic acids of the formula CnF2n +1-C(= O)OH where n = 8, 9, 10, 11, 12, or 13 (C9-C14 PFCAs) including their salts and any combination thereof.
These are the six C9-C14 PFCAs namely:
C9- PFCA - PFNA, EC Nr. 206-801-3 CAS Nr. 375-95-1
C10-PFCA - PFDA, EC Nr. 206-400-3 CAS Nr. 335-76-2
C11-PFCA - PFUnDA EC Nr. 218-165-4 CAS Nr. 2058-94-8
C12-PFCA - PFDoDA EC Nr. 206-203-2 CAS Nr. 307-55-1
C13-PFCA - PFTrDA EC Nr. 276-745-2 CAS Nr. 72629-94-8
C14-PFCA - PFTeDA EC Nr. 206-803-4 CAS Nr. 376-06-7
and their salts,
- Any C9-C14 PFCA-related substance having a perfluoro group with the formula CnF2n +1- directly attached to another carbon atom, where n = 8, 9, 10, 11, 12, or 13, including their salts and any combinations thereof.
These are the related substances which can degrade or be transformed to the C9-14 acids. PFCA-related substances are substances that, based upon their structural formulae, are considered to have the potential to degrade or be transformed to C9-14 perfluorocarboxylic acids (linear and/or branched).
- e.g. Ammonium nonadecafluorodecanoate

and 2,2,3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,12,12,12-docosafluoro-11-(trifluoromethyl)- dodecanoyl fluoride

however, note that the substance methyl nonacosafluoropentadecanoate has a perfluoro group CnF2n+1- directly attached to another carbon atom however the perfluorinated chain (C14F29-) does not fall within the range n=8, 9, 10, 11, 12 or 13 and is outside the scope of the restriction. The nomenclature CnF2n+1- means any branched or linear perfluorinated alkyl moiety containing carbon atoms on which all the H substituents have been replaced by F atoms. The `-´ represents ‘bonded to’ and any group can be bonded to the CnF2n+1- moiety, including for example iodine. If CnF2n+1- is directly attached to another carbon atom it is out of scope.
3. Any C9-C14 PFCA-related substance having a perfluoro group with the formula CnF2n +1- that it is not directly attached to another carbon atom, where n = 9, 10, 11, 12, 13 or 14 as one of the structural elements, including their salts and any combinations thereof.
An Example of this group is: 1,1,1,2,2,3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11-tricosafluoro-11-iodo-undecane:

The following substances are excluded:
4. Substances with formula CnF2n +1-X, where X = F, Cl, or Br
where n = 9, 10, 11, 12, 13 or 14, including any combinations thereof.
These are perfluorinated substances with a halogen attached and as such are a different group of substances that are not degraded to PFCAs. Iodine is not excluded by this group because it is the starting point for the telomerisation process.
5. Substances with formula CnF2n +1-C(= O)OX' where n> 13 and X'=any group, including salts.
The nomenclature X’ means any possible functional group.
This exclusion is for substances that contain perfluoro groups having higher carbon numbers than those mentioned under paragraph 1 and are as such not covered by the restriction because only C9-C14 PFCAs have been identified as PBT or vPvB substances.
Both linear and branched C9-C14 perfluorocarboxylic acids are included in the scope of restriction entry 68. Polyfluorinated (i.e. partially fluorinated) substances containing a structural element with a sufficiently long perfluorinated moiety are included within the scope of the restriction because they degrade to perfluorinated (fully fluorinated) C9-C14 PFCAs e.g. 8:2 FTOH:

Polyfluorinated substances containing other partially fluorinated structural elements such as the substance below are not included within the scope of the restriction because they do not contain a structural element with a sufficiently long perfluorinated moiety (e.g., 2,2,3,3,4,4,5,6,6,7,7,8,8,9,9,9-hexadecafluorononyl methacrylate:

It should be noted that if a substance contains structural elements both inside and out of scope, then the substance is still within the scope.
More information is available in the Final background document accessible on the ECHA website in the Registry of restriction intentions until outcome section.
This guideline clarifies how users of NMP can ensure their compliance with the derived no-effect levels (DNELs) established in the restriction on 1-methyl-2-pyrrolidone (NMP) in entry 71 of Annex XVII to the REACH Regulation (EU) No 1907/2006. The guideline explains how to proceed when there are mandatory DNELs in addition to the occupational exposure limit values (OELs). It clarifies what the user needs to do to adequately control the risks. Several examples of good practice illustrate the type of equipment some companies have in place to control inhalation and dermal exposure in various tasks such as transfers, maintenance and sampling. In addition, examples of monitoring methods, including biological monitoring methods, are described. The document has been prepared in close cooperation with the industry stakeholders and endorsed by the Member States authorities.
In your language:
The use of NMP (as such or in a mixture) during the production of articles is covered by the restriction. This means that you have to apply the conditions set by the restriction in your production operations. More information on how to do that is available in the NMP guideline.
The use of articles containing NMP is not considered as use of the substance (as such or in a mixture), and thus is not subject to the restriction. This means that your customers using the article you produce do not need to comply with the conditions set by the restriction.
However, it is important to note that NMP is listed in the Candidate List and specific obligations apply to its presence in articles. Similar duties are also set in the revised Waste Framework Directive 2008/98/EC. For more information see SCIP database.
Last but not least, if you are not sure how to differentiate articles from substances or mixtures, the ECHA guidance on requirements for substances in articles, Appendix 4 will help you.
Appendix 12 of Annex XVII which is referred to in entry 72 includes five phthalates, i.e. 1,2-benzenedicarboxylic acid; di-C 6-8-branched alkylesters, C 7-rich, bis(2-methoxyethyl) phthalate, diisopentylphthalate, di-n-pentyl phthalate (DPP) and di-n-hexyl phthalate (DnHP).
Clothing and related accessories, textiles other than clothing which under normal or reasonably foreseeable conditions of use come into contact with human skin to an extent similar to clothing and footwear cannot contain a total concentration equal or greater than 1000 mg/kg of the phthalates specified in the Appendix 12, considered individually or in combination with other phthalates listed either in i) Appendix 12, or ii) elsewhere in Annex XVII, if classified as carcinogenic, mutagenic or toxic to reproduction (CMR), category 1A or 1B in Part 3 of Annex VI to Regulation (EC) No 1272/2008.
Examples of other phthalates in Annex XVII include those listed in entry 51 (bis (2-ethylhexyl) phthalate (DEHP), benzyl butyl phthalate (BBP), dibutyl phthalate (DBP) and diisobutyl phthalate (DIBP)). These substances are classified as toxic to reproduction category 1B, and therefore fulfil the criteria of the restriction. Thus they cannot be present in the clothing and related accessories, other textiles and footwear placed on the market if, in combination with the phthalates listed in Appendix 12, the total phthalate concentration, measured in homogeneous material, is equal to or greater than 1000 mg/kg. Note also that Appendices to entries 28 to 30 provide lists of substances classified as CMRs, category 1A or 1B to which the restrictions apply. The above considerations are relevant also to the phthalates included in those Appendices.
Restriction entry 73 concerns ‘(3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctyl) silanetriol and any of its mono-, di- or tri-O-(alkyl) derivatives (hereafter referred as TDFAs)’
The restriction targets spray products for supply to the general public (e.g. aerosol dispensers, pump and trigger sprays and mixtures marketed for spray application) containing mixtures of TDFAs and organic solvents.
A general formula for the targeted group of substances can be given as:

In theory, variations in the alkoxy chain can give rise to a very large number of parent compounds. It is important to note, however, that the restriction applies to all substances which are covered by the general formula in order to avoid substitution to other polyfluorooctyl alkoxysilanes.
Examples of substances targeted by the restriction:

More information is available in the Final background document accessible on the ECHA website in the Registry of restriction intentions until outcome section.
Restriction entry 74 applies to ‘Diisocyanates, with generic formula O=C=N-R-N=C=O, with R being an aliphatic or aromatic hydrocarbon unit of unspecified length’. This means that the group R in this entry does not contain urethane, urea, uretdione, biuret, allophanate or isocyanurate linkages (i.e., the diisocyanate entity is not the result of prepolymerisation of a parent diisocyanate). The restriction covers industrial and professional uses of Diisocyanates (as substances on their own, as constituents of other substances or in mixtures) in concentration ≥0.1% by weight. As a result, oligomers and prepolymers that contain ≥0.1% (by weight) of the diisocyanates that meet the above definition (of which a non-exhaustive list is shown below), would still be in scope of the restriction.
Examples of diisocyanate substances covered by restriction entry 74:

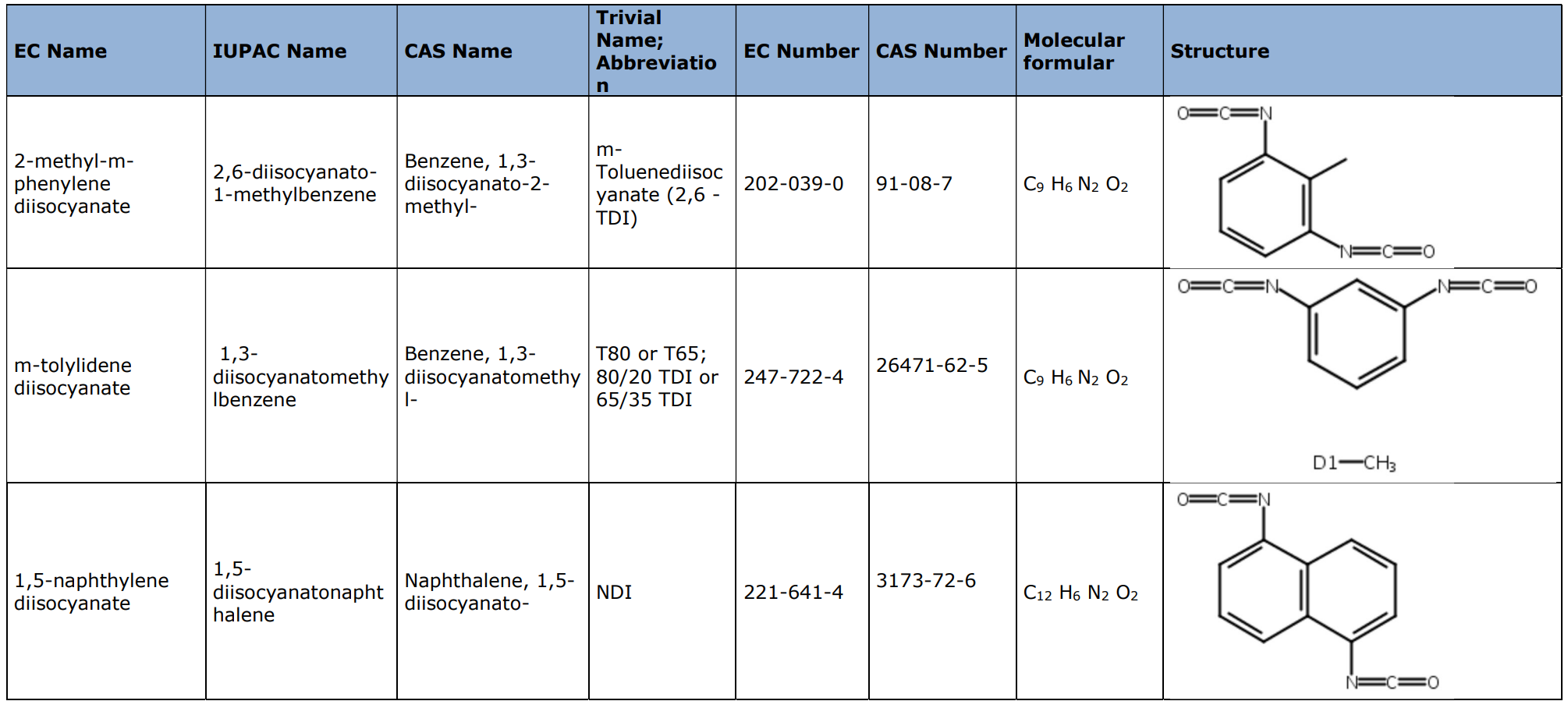

More information is available in the Final background document accessible on the ECHA website in the Registry of restriction intentions until outcome section.
Paragraph 4 of the restrictions indicates that the “training shall be conducted by an expert on occupational safety and health with competence acquired by relevant vocational training”. Restriction entry 74 does not contain additional education requirements (e.g., University degree on Chemistry or Toxicology) for trainers. Trainers must be able to demonstrate their qualification as expert on occupational safety and health and, in addition, to have acquired specific competence in the fields relevant for the restriction through vocational trainings. The qualifications of the experts should comply with specific provisions on educational and professional requirements of experts on occupational safety and health as required by the applicable legislation, including those established by the Member States.
There are no provisions in restriction entry 74 on how to deliver the training, therefore it can be assumed that the content can be provided also online through pre-prepared recorded and/or interactive training sessions. However, a qualified trainer (as described above) is needed to provide responses to possible questions by participants during or after the training and to verify and validate possible final tests aiming to verify if the content of the training has been understood by participants. It should be considered that the scope of the restriction is to ensure that workers handling diisocyanates have a thorough understanding of the risks and of the health and safety measures to adopt to control them (including in emergency situations).
As outlined in paragraph 1 of the restriction entry 74 of Annex XVII to REACH,
Diisocyanates,
1. Shall not be used as substances on their own, as a constituent in other substances or in mixtures for industrial and professional use(s) after 24 August 2023, unless:
a) the concentration of diisocyanates individually and in combination is less than 0,1 % by weight, or
b) the employer or self-employed ensures that industrial or professional user(s) have successfully completed training on the safe use of diisocyanates prior to the use of the substance(s) or mixture(s).
It is further outlined in paragraph 5 of the restriction that:
The training referred to in point (b) of paragraph 1 shall include the instructions for the control of dermal and inhalation exposure to diisocyanates at the workplace without prejudice to any national occupational exposure limit value or other appropriate risk management measures at national level. Such training shall be conducted by an expert on occupational safety and health with competence acquired by relevant vocational training.
That training shall cover as a minimum:
a) the training elements in point (a) of paragraph 5 for all industrial and professional
use(s).
b) the training elements in points (a) and (b) of paragraph 5 for the following uses: handling open mixtures at ambient temperature (including foam tunnels), spraying in a ventilated booth, application by roller, application by brush, application by dipping and pouring, mechanical post treatment (e.g. cutting) of not fully cured articles which are not warm anymore, cleaning and waste, any other uses with similar exposure through the dermal and/or inhalation route.
c) the training elements in points (a), (b) and (c) of paragraph 5 for the following uses: handling incompletely cured articles (e.g. freshly cured, still warm), foundry applications, maintenance and repair that needs access to equipment, open handling of warm or hot formulations (> 45 °C), spraying in open air, with limited or only natural ventilation (includes large industry working halls) and spraying with high energy (e.g. foams, elastomers and any other uses with similar exposure through the dermal and/or inhalation route.
The text of the restriction states that the employer (or self-employed worker) has full responsibility that industrial and professional workers have successfully completed a training on the safe use of diisocyanates before they use these substances (or mixtures).
Suppliers shall ensure that the recipients of substance(s) or mixture (s) are provided with the training materials and courses (paragraph 7). The restriction does not specify who has to prepare that material, but the material provided by the supplier must comply with paragraphs 4 and 5 of the entry and take into consideration the specificity of the products supplied, including composition, packaging, and design. Therefore, input of the manufacturer/producer may be relevant.
Furthermore, the restriction indicates that the training has to be conducted by an expert on occupational safety and health with competence acquired by relevant vocational training. Minimum requirements for the training are also indicated in the entry.
The employer is responsible to select the training provider, who is responsible to provide the training covering as a minimum the elements laid down in paragraph 5 of the restriction. The employer will have also the duty to ensure that the provider is qualified to conduct the training and to verify that the training material provided by the supplier covers as a minimum the topics indicated in points a) and b) and, where applicable c), of paragraph 5 of the restriction and that the training effectively provided by the expert covers such topics.
The restriction entry 74 sets conditions on the use of diisocyanates as substances on their own, as a constituent in other substances or in mixtures for industrial and professional use as well as on placing them on the EU/EEA market. The restriction entry is available in the document ANNEX XVII to REACH – Conditions of restriction.
The restriction does not apply to articles as defined under REACH. For more information, see the ‘Guidance on requirements for substances in articles’.
The supplier of the substance or mixture is responsible for providing the required information and that the packaging contains the required statement as per paragraph 2(b) of restriction entry 74 of Annex XVII to REACH.
Under the REACH regulation, a supplier is defined as “any manufacturer, importer, downstream user or distributor placing on the market a substance, on its own or in a mixture, or a mixture” (Art. 3(32) of REACH). Furthermore, ‘placing on the market’ means supplying or making available, whether in return for payment or free of charge, to a third party. Import is deemed to be placing on the market (Art. 3(12) of REACH). Therefore, any supplier, including the manufacturer of these substances, the formulator of mixtures containing these substances and a distributor down the supply chain, needs to ensure the provision of the correct information and required statement on the packaging of the substance or mixture they place on the market, even if the substance or mixture was manufactured or formulated before 24 February 2022.
Restriction entry 75 of Annex XVII to REACH covers different categories of substances that are considered to pose risks to human health if they are used for tattooing purposes. The substances restricted for use in tattoo inks and permanent make up include:
1. Substances with the following harmonised classifications:
- carcinogenic and mutagenic (CM), categories 1A, 1B and 2 if present in mixtures for use for tattooing purposes in a concentration ≥0,00005 % by weight
- reproductive toxicants (repro), categories 1A, 1B and 2 and skin sensitisers (SS) Cat. 1, Cat. 1A and Cat. 1B if present in mixtures for use for tattooing purposes in a concentration ≥0,001 % by weight
- Substances with harmonised classification as skin irritant (Cat. 2), skin corrosive (Cat. 1, Cat. 1A, 1B, 1C), eye irritant (Cat. 2) or serious eye damaging (Cat. 1) if present in mixtures for use for tattooing purposes in concentration ≥ 0,1 % by weight (if the substance is used only as a pH regulator) or ≥ 0,01 % by weight, in all other cases;
NOTE: substances with harmonised classification as carcinogen, mutagen and reproductive toxicant category 1A, 1B or 2 for effects due only to inhalation exposure are excluded.
2. Substances in the scope of Regulation (EU) 1223/2009 - Cosmetic Products Regulation (CPR) – present in mixtures for use for tattooing purposes in concentration ≥0,00005 % by weight i.e.:
- Substances listed in Annex II of CPR Regulation (list of substances prohibited in cosmetic products)
- Substances listed in Annex IV of CPR Regulation
- Restricted due to condition of use of one or more of this kind specified in column g of Annex IV: “Rinse-off products”; “Not to be used in products applied on mucous membranes”; “Not to be used in eye products”
- the substance is present in the mixture in a concentration, or in some other way, that does not accord with the conditions specified in column h or i of Annex IV.
3. Substances included in Appendix 13 of Annex XVII to REACH
The REACH restriction does not contain a positive list of substances that can be used in tattoo inks and permanent make-up. Substances not included in the categories indicated in restriction entry 75 of Annex XVII to REACH above the limits specified therein or in any other applicable entry (e.g., entries 28-30 ban the placing on the market, or use, for supply to the general public of substances with harmonised classification as carcinogen, germ cell mutagen or reproductive toxicant, categories 1A or 1B listed in appendices 1 to 6 of Annex XVII to REACH) are allowed to be used in tattoos.
The following substances are excluded from restriction entry 75:
1. Substances classified as carcinogens or mutagens in Categories 1A, 1B and 2 only with the hazard statements H350i (May cause cancer by inhalation), H351i (Suspected of causing cancer by inhalation), H340i (May cause genetic defects via inhalation) and H341i (Suspected of causing genetic defects by inhalation)
2. Substances classified for reproductive toxicity (repro) categories 1A, 1B and 2 only with the hazard statement H340(inhalation) (May cause genetic defects via inhalation) and H341(inhalation) (Suspected of causing genetic defects by inhalation)
3. Substances that are gases at temperature of 20 °C and pressure of 101,3 kPa, or generate a vapour pressure of more than 300 kPa at temperature of 50 °C., with the exception of formaldehyde (CAS No 50-00-0, EC No 200-001-8)
The following substances can still be used in tattoos until 4th of January 2023:
- Pigment Blue 15:3 (CI 74160, EC No 205-685-1, CAS No 147-14-8);
- Pigment Green 7 (CI 74260, EC No 215-524-7, CAS No 1328-53-6
Mixtures placed on the market for use for tattooing purposes need to be marketed with the statement ‘Mixture for use in tattoos or permanent make-up’. The mixture must contain information indicated in Paragraph 7 of the restriction entry.
More information is available in the Final background document accessible on the ECHA website in the Registry of restriction intentions until outcome section.
Operators in the EU/EEA need to comply with the conditions specified in any relevant restriction entry that applies to substances placed on the market in a product or used in the EU/EEA market.
This means that manufacturers, importers and downstream Users (DU) of the substances listed in the restrictions must comply with the provisions of Entry 75. For instance, a tattoo artist who is formulating a mixture needs to comply with the relevant obligations of this restriction as he/she is considered as a downstream user. For further details, see Q&A 171.
The enforcement of REACH restrictions is a responsibility of each EU Member State, as well as the members of the EEA (Norway, Iceland and Liechtenstein). They must ensure that there is a system of official controls and lay down legislation specifying penalties for non-compliance with the provisions of REACH. For further details, see Q&A 3.
Many substances have multiple harmonised classifications and some of them are also included into Annex II or Annex IV of the CPR Regulation. Concentration limits specified in entry 75 of annex XVII to REACH may differ for different category of substances. If a substance falls into more than one of the categories of substances restricted under Entry 75 of Annex XVII to REACH, the lowest of the concentration limits applicable to each category has to be taken into account.
As an example, formaldehyde has a harmonised classification as Carc 1B, Muta 2 , and a harmonised classification as Skin Sens. 1. The following concentration limits are established in entry 75:
- 0,00005 % by weight for substances with harmonised classification as Carc 1 B ad Muta 2
- 0,001 % by weight for substances with harmonised classification as Skins sens 1.
In the case of Formaldehyde the limit of 0,00005% by weight applies in light of its classification as Carc 1B/Muta 2.
Restriction entry 75 applies from the date of the entry into application of the harmonised classification for substances that receive a Harmonised Classification which falls into the scope of the entry (i.e. CM 1A, 1B or 2 Repro 1A, 1B or 2, Skin Sens 1, 1A or 1B, Skin Corr. 1, 1A, 1B or 1C, Skin Irritant 2, serious eye damage category 1 or eye irritant category 2.
If a substance is included into Annex II or Annex IV of the CPR Restriction entry 75 applies 18 months from the date of inclusion in the relevant Annex.
Information on suitable analytical methods to verify compliance to the provisions in entry 75 is provided in the Annex to the Background Document, Appendix D.2. However, national enforcement authorities of Member States may have developed additional test methods for tattoo inks.
- The risk management measures for the identified uses with regard to human health and the environment are to be summarised in section 8 (and 7). This includes consumer related measures communicated to a downstream user producing consumer preparation or articles. Also the relevant Derived No-Effect Levels (DNELs) and Predicted No-Effect Concentrations (PNECs) should be presented here.
- The information on physicochemical properties, toxicology and eco-toxicology in the SDS is to be updated in line with the information requirements of Annex VI to XI of the REACH Regulation.
- The results of the PBT and vPvB assessment are to be presented in section 12.
- The information on uses advised against in section 16 of the SDS may need to be updated depending on the outcome of the manufacturer's Chemicals Safety Assessment (CSA).
- Where Exposure Scenarios (ES) are developed as a result of conducting a chemical safety assessment in accordance with Article 14 of the REACH Regulation they must be annexed to the SDS and thereby be appropriately passed down the supply chain. The information on uses of the substance in section 1.2 of the SDS must be consistent with the short titles of the ES in the annex, indicating which uses are covered by the ES.
- Since REACH includes a requirement to include the waste disposal considerations into the manufacturer's chemicals safety assessment, section 13 of the SDS may need to be updated with substance specific waste management advice as contained in the ES.
According to Article 31(5) of the REACH Regulation, the safety data sheet (SDS) shall be supplied in an official language of the Member State(s) where the substance or mixture is placed on the market, unless the Member State(s) concerned provide otherwise. Placing on the market means supplying or making available, whether in return for payment or free of charge, to a third party. Import shall be deemed to be placing on the market (Article 3(12) of the REACH Regulation).
The above also applies to exposure scenarios, which are a part of an SDS. A document listing the languages required for safety data sheets and labels within the EU is available at http://echa.europa.eu/documents/10162/13562/languages_required_for_labels_and_sds_en.pdf
For a substance or mixture requiring a Safety Data Sheet (SDS) according to Article 31 of the REACH Regulation, Annex II of REACH requires that the registration number assigned in accordance with Article 20 of REACH be given in the SDS when it is available. Article 31(9) of REACH gives specific occasions on which an updated SDS should be supplied without delay. Receipt of a registration number per se is not listed as one of these occasions. However as the assignment of a registration number is of major potential interest to downstream users of the substance it may be recommendable to send existing customers an updated SDS either immediately or on the next supply of the substance or of a mixture containing it. The SDS should of course be updated to incorporate the registration number(s) for customers who are going to receive the substance or mixture for the first time. Note in particular that the final sentence of Article 31(9) requires that "any updates following registration shall contain the registration number". Please note that there are detailed provisions in Regulation (EU) No 453/2010 amending Annex II to the REACH Regulation regarding when the part of the registration number referring to the individual registrant of a joint submission (the last four digits of the original full registration number) may be omitted by a supplier who is a distributor or a downstream user.
Similarly, Article 32(1)(a) of REACH indicates that when registration numbers have to be communicated to customers according to Article 32 (Communication duties for substances and mixtures not requiring an SDS) the registration number, if available, should be supplied. The occasions on which an update without delay is required are given in Article 32(3) of REACH. Again, receipt of a registration number per se is not listed as one of these occasions. For similar reasons it may be deemed to be desirable to nonetheless send updated information. Note again that the last sentence of Article 32(3) of REACH also requires that "any updates following registration shall contain the registration number". Further guidance can be found in section 6.1.2-'Provide other information to customers' in the Guidance on Registration available on the ECHA website at: http://echa.europa.eu/guidance-documents/guidance-on-reach.
The provisions of Articles 31 and 32 of REACH apply irrespectively of whether the relevant registration deadline is still to come or has already elapsed.
There is no obligation under REACH for suppliers of substances and mixtures meeting the criteria in Article 31 to provide a SDS to their non-EU customers. Article 31(1) refers to "recipients of the substance or mixture". Article 3(34) of REACH defines a "recipient of a substance or a mixture" as being downstream user or a distributor being supplied with a substance or a mixture. Both downstream users and distributors are, in line with their respective definitions in Article 3(13) and 3(14), natural or legal persons established within the Community. The obligation of Article 31 of REACH to provide an SDS therefore applies only to the recipients of the substance or mixture established in the EU.
However, it is notable that the obligation to provide a REACH compliant SDS to non-EU customers, in the context of export, may arise pursuant to other pieces of legislation. For example, Article 16(3) of Regulation (EC) No 689/2008 concerning the export and import of dangerous chemicals, implementing the Rotterdam Convention within the EU, requires companies exporting certain hazardous chemicals within the scope of this Regulation to provide a REACH compliant SDS when exporting them outside the EU.
No, it is not. Regarding section 2 of the SDS, either the full wording of the hazard classes or the hazard class and category code(s) may be used. If the full wording is used, it needs to be in the language of the SDS. If the hazard class and category code(s) are used, it is important to note that the abbreviations given for each hazard class are actually codes which cannot be translated. The codes must thus remain as they are given in Annexes VI and VII to CLP. If codes, other abbreviations and acronyms are used, their full text and explanation must be given in section 16 of the SDS, in the language of the SDS.
For mixtures, the codes as given in Annexes VI and VII to CLP can be used in section 3.2.3. Again, section 16 needs to contain the full wording.
No, manufacturers or importers do not have the obligation to inform downstream users that they have submitted a notification to the C&L Inventory. Furthermore, there is no need for downstream users to receive confirmation from upstream suppliers that substances have been notified to the C&L Inventory in order to continue the use of the substances in their own products. Similar to the REACH pre-registration number, the C&L notification number is for internal use for the importer/manufacturer as receipt/proof of notification. It does not need to be communicated to the DU/distributor.
A notification number cannot be considered as an identifier according to Article 18 CLP and it is not the inventory reference number published in the C&L Inventory.
Substances e.g. pre-registered, inquired or notified to the C&L Inventory with only a CAS number or without any numerical identifier are automatically assigned a list number. In contrast to the EINECS, ELINCS and NLP entries, the list numbers and the list inventory are not based on a legal act or requirement, and they have not been published in the Official Journal of the European Union. Therefore, the list numbers do not have the same significance as EC numbers but have only the numerical format in common. Most importantly, the vast majority of list numbers and their connected substance identification have never been checked for correctness, validity or whether the conventions outlined in the Guidance for Identification and naming of substances under REACH have been kept.
Therefore, industry is advised not to use list numbers in their documents.
However, when a supplier wishes to include a list number on a document, e.g. Safety Data Sheet, it shall be clearly indicated that this number is not an EC number and has no legal significance.
Distributors/formulators can truncate the registration number (omit the last four digits) of their suppliers' registration numbers in the SDS in accordance with points 1.1 and 3.2.4 of Annex II of REACH.
The first part of the registration number is the same for a given substance if the registrants have jointly submitted their registrations. Therefore, only one truncated registration number needs to be included. However, if a registrant has not registered in the context of the joint submission, this registration number is different. Therefore, all relevant (truncated) registration numbers should be mentioned in the SDS.
For registered substances for which a Chemical Safety Report (CSR) is required, the information appearing in section 1.2 of the SDS needs to be in line with the identified uses in the CSR (under REACH, the definition of use goes beyond the chemical function) and the Exposure Scenario (ES) annexed to the SDS. Intuitive ES titles can be reported in section 1.2. When the use descriptor system is used in the ES, it is advised that, in section 1.2, the uses of the substance are described in a generic wording while remaining consistent with that of the use descriptor system. Reporting the use descriptor codes in section 1.2 is not recommended as it may lead to lengthy lists. The Process and Product Categories in the use descriptor system can be used as an indication.
For registered substances for which a CSR is not required (between 1-10 tonnes/year), substances not yet registered or not subject to registration (e.g. below 1 tonne/year or listed in Annex IV or V) and mixtures, the intended uses known to the supplier need to be indicated, with a brief description of what the substance or mixture is intended to do, such as "flame retardant in textile fibres", "antioxidant in paints, cosmetics, detergents", etc.
For downstream users in a supply chain who must generate their own safety data sheets for products procured from within the EEA, the main source of information should be the supplier. Using information from the supplier will ensure that any specific, detailed information is reviewed and included as appropriate.
In addition, use may be made of publicly available information that is not restricted by copyright or any confidentiality protections. It should be noted that, as stated in ECHA's legal notice, reproduction or further distribution of the information from the registration dossiers may be subject to copyright protection and that the use of the information without obtaining the permission from the owner(s) of the respective information might violate the rights of the owner.
Moreover, the legal notice emphasises that the Agency cannot guarantee the correctness of the information in the databases and REACH does not permit the Agency to make modifications to the data provided by the owner(s) of the respective information.
It should be noted that the supplier of the Safety Data Sheet is responsible for the accuracy of its contents in all cases.
Where an Exposure Scenario (ES) is developed as a result of conducting a chemical safety assessment as required by Article 14 or 37(4) of the REACH Regulation, it must be annexed to the safety data sheet (SDS), provided it is relevant for the customer.
According to Article 31(9) a supplier shall update his SDS without delay as soon as new information which may affect the risk management measures or new information on hazards becomes available. An ES is considered to be such new information, which normally triggers the need to update a SDS as soon as it becomes available. The SDS with annexed ES resulting from this update has to be provided without delay to all customers who have been supplied with the particular substance or mixture within the preceding 12 months. This provision applies since the entry into force of the REACH Regulation and irrespectively of whether the substances are registered or not.
Where the information in an ES that becomes available does not affect the risk management measures and the ES contains no new information on hazards, the SDS does of course not have to be updated.
Yes, if the conditions of Article 31(1) of the REACH Regulation are fulfilled, the supplier of a biocidal product that is a substance or a mixture must provide the recipient with a safety data sheet.
When a formulator produces a biocidal product, the relevant safety measures (or restrictions on use) indicated in the product authorisation should be communicated down the supply chain (with the SDS, where required).
An active substance which is either approved under the BPR at EU level or part of the BPR Review Programme, is considered as registered under REACH only for the use in a biocidal product.
There is no requirement to attach an exposure scenario to the SDS for the active substance used in a biocidal product. REACH exposure scenarios may, nevertheless, be received for co-formulants in the biocidal product. The formulator needs to take into account the relevant information in these exposure scenarios in the SDS, where required.
(See also BPR Q&A 908).
Yes, the SDS format can be used. Suppliers, who do not have to supply an SDS may be obliged to provide certain information in accordance with Article 32 of the REACH Regulation or they may chose to provide a SDS on a voluntary basis. They may use the SDS format to provide such information. In such cases, it is recommended that it is clearly stated that the SDS is not provided pursuant to Article 31 of REACH, but to facilitate the communication of information.
The suppliers of a substance or a mixture may also choose to provide information in the safety data sheet format even if they are not obliged to provide any information under Articles 31 or 32 under REACH. In this case, they should also clarify that the SDS is not provided pursuant to Article 31 of REACH, and then explain why they provide it.
Among possible solutions would be the addition to the relevant SDS of a phrase such as ‘A safety data sheet is not required for this product under Article 31 of REACH’.Yes it can if the information provided in the SDS fulfills the requirements of Annex II of REACH for each of those substances or mixtures. However, this condition can only be fulfilled in cases of minor variations of a substance or a mixture, for example for minor concentration changes of impurities or components, due to which the hazard profile does not change. A single SDS cannot be used for totally different substances or mixtures.
While it is not legally required under Annex II of the REACH Regulation to include EUH statements for substances in a mixture in section 3.2 of the SDS, it would be advisable and good practice to include those substances assigned a physical and health property as set out in Part 1 of Annex II and Table 1.2 of Annex VII as these were translated from risk phrases assigned under Directive 67/548/EEC e.g. EUH032. Furthermore, it would also be advisable and good practice to include these EUH statements in Section 2.1 and 2.2 of the SDS. For other EU hazard statements it is only necessary to include them in Section 2.2 of the SDS.
Note: COM may need to consider including provision for EU hazard statements relating to supplemental hazard information taken over from Directive 67/548/EEC in the next update of Annex II of REACH.
Yes, you do. The obligation to mention any European or national occupational exposure limits (OELs) is not connected to the physical form or to the classification of the mixture, or even to the fact that the mixture can be considered as a special mixture. Where an OEL exists for any constituent substances, you must indicate these in Section 8 of the safety data sheet (SDS).
An SDS must enable employers to determine whether any hazardous chemical agents are present in the workplace and to assess any risk to the health and safety of workers arising from their use.
It should also be taken into account that a user might use the paste in a way that it undergoes further processing.
You do not need to indicate whether the substance requires a safety data sheet. REACH-IT will determine this and consequently whether a confidentiality claim needs to be invoiced or not. For determining whether the substance requires a safety data sheet, REACH-IT is verifying in the registration dossier if the conditions set out in Article 31 of REACH are met:
- the substance is classified as hazardous according to the CLP Regulation2; and/or
- the substance is PBT or vPvB; and/or
- the substance is listed on the candidate list;
- and it concerns a full registration or a registration for a transported isolated intermediate.
There can be several reasons why the SDS for a substance does not include exposure scenarios:
- Substance is not registered yet;
- Substance is registered below 10 tonnes per year;
- Substance is registered as an intermediate;
- The SDS is provided on a voluntary basis;
- The SDS is required, but the registrant was not obliged to carry out an exposure assessment according to Article 14(4) REACH;
If the SDS you receive is for a mixture containing one or more registered substances, the reason you did not receive exposure scenarios could be that the relevant information from the exposure scenario is incorporated in the main body of the SDS or in an annex for the mixture (also called SUMI, Safe Use of Mixture Information).
Follow the safety advice in the SDS unless it is inappropriate. In such an event, you are obliged to inform your supplier of the reason why you consider their advice to be inappropriate. Contact your supplier if you think that an exposure scenario should have been provided.
Member States endorse national provisions defining controls and sanctions for non-compliance with REACH. We recommend you to contact the relevant enforcement authorities in your country for information on the national control procedures in place. You can also contact the customs authorities and the national helpdesk for further information.
Yes, because besides registration there are several obligations under REACH that apply irrespective of tonnage. These include restrictions, authorisation and communication in the supply chain (such as the provision of safety data sheets). The one tonne per year threshold applies to registration only.
To identify your obligations, we recommend you to use the Navigator tool.
Active substances for use in biocidal products are regarded as already registered, as biocidal products and their active ingredients are covered by Regulation (EU) 528/2012 (Biocidal Products Regulation). However, several conditions have to be fulfilled to benefit from the exemption. These conditions are laid down in Article 15(2) of the REACH Regulation and explained in section 2.2.4.1- 'Substance for use in biocidal products' of the Guidance on registration http://echa.europa.eu/guidance-documents/guidance-on-reach.
Active substances for use in plant protection products (PPPs) are regarded as registered as the plant protection products and their active ingredients are covered by Regulation (EC) 1107/2009. Please note that even though co-formulants are mentioned in Article 15(1) of the REACH Regulation, currently they do not meet the conditions laid down in this Article. Therefore co-formulants used in plant protection products do not qualify for the exemption and are not regarded as registered. This is further explained in section 2.2.4.2- 'Substance for use in plant protection products' of the Guidance on registration http://echa.europa.eu/guidance-documents/guidance-on-reach.
It is important to note, that only the quantities of the active substance for use in biocidal products and for use in PPPs are considered registered under REACH. Thus, if the substance is not used as an active ingredient in a biocidal product or a PPP, then the exemption would not apply to this other use and the quantity of the substance for the non-biocidal or non-PPP use would have to be registered.
REACH is an EU Regulation that directly applies in all Member States of the European Union. REACH is of EEA (European Economic Area) relevance as it has been incorporated into the Agreement on the European Economic Area. This means that REACH also applies in Iceland, Liechtenstein and Norway.
Substances imported into the EEA from Switzerland (a non-EU country belonging to the European Free Trade Association but not to the EEA) are treated under REACH in the same way as substances imported from any other non-EEA country.
The Member States are best placed to explain how REACH applies to their territories (autonomic areas or overseas territories). We therefore recommend contacting the national REACH helpdesk of the relevant country to clarify specific questions in this regard.
Companies established outside of the EU/European Economic Area (EEA) do not have obligations under REACH. It is the EU importer that sources substances from the non-EU/EEA company who must comply with REACH.
Non-EU/EEA manufacturers and formulators exporting substances to the EU can (but are not obliged to) appoint an "only representative" to fulfil the obligations of importers. More guidance on only representatives can be found in our Q&A section on Only Representatives and in the Guidance on registration section 2.1.2.5- "Only representative of a non-EU manufacturer".
For further information, see also the webpage concerning the non-EU companies.
An importer is any natural or legal person established within the Community who is responsible for import (Article 3(11) of the REACH Regulation). Import means the physical introduction into the customs territory of the Community (Article 3(10)).
The scope of the customs territory of the Community is defined in the relevant Community legislation on customs that should be consulted for further information. [Regulation (EU) No 952/2013 of the European Parliament and of the Council of 9 October 2013 laying down the Union Customs Code].
It is important to note, however, that the REACH Regulation and the Customs legislation are independent from each other.
The import of a substance on its own, in mixtures or in articles manufactured or produced outside the European Union is considered placing the substance, mixture or article on the Union market.
The responsibility for import depends on many factors such as who orders, who pays, who is dealing with the customs formalities, but this might not be conclusive on its own.
Example
A sales agency established in the EU that acts as an intermediary, i.e. they transmit orders from buyers to non-EU suppliers (and are paid for this service). However, they take no responsibility for the goods or the payment of the goods and do not own the goods at any stage. In this case, the sales agency is not considered to be an importer for the purposes of REACH. The sales agency is not responsible for the physical introduction of the goods.
In many instances, this will be the ultimate receiver of the goods (the consignee) who is the legal entity responsible for importing the goods.
For further information and examples see the Guidance on registration, chapter 2.1.2.4 "Who is responsible for registration in case of import?".
Under REACH, manufacturing means producing or extracting substances in their natural state. It is a case-by-case decision to establish the extent to which the different steps in producing the substance are covered by the definition ‘manufacturing'.
Example
A company that purchases registered substances from within the EU and then formulates these into mixtures (e.g. paints) would be regarded as a downstream user under REACH.
In layman's terms, this company might be considered to be a manufacturer of paints. However, within the context of REACH, the company would not be a manufacturer of a substance and so would have no registration obligations for these substances. For further information see the Guidance on registration. Examples on manufacturing of intermediates available in the Guidance on intermediates and Practical Guide 16.
You should register if you are the legal entity established in the EU who is responsible for importing.
The responsibility for importing depends on many factors such as:
- Who orders?
- Who pays?
- Who is dealing with the customs formalities?
However, this might not be conclusive on its own.
Example
A sales agency established in the EU that acts as an intermediary, i.e. they transmit orders from buyers to non-EU suppliers (and are paid for this service). However, they take no responsibility for the goods or the payment of the goods and do not own the goods at any stage. In this case, the sales agency is not considered to be an importer for the purposes of REACH. The sales agency is not responsible for the physical introduction of the goods.
In many instances, this will be the ultimate receiver of the goods (the consignee) who is the legal entity responsible for importing the goods.
For further information and examples see chapter 2.1.2.4 "Who is responsible for registration in case of import?" in the Guidance on registration:
If substances are in temporary storage with a view to re-exportation and remain under customs supervision, they are not subject to REACH.
To rely on this exemption, you need to document that the following conditions are fulfilled:
1.The substances are put in a free zone or free warehouse as defined under customs legislation or placed under another relevant customs procedure (transit procedure, temporary storage).
2.The substances are kept under the supervision of the customs authorities.
3.The substances do not undergo any form of treatment or processing during their stay in the EU. A free zone or a free warehouse in the EU territory is part of the EU.
For further information see the Guidance on registration, chapter 2.2.2.2 ‘Substances under customs supervision’.
Annex V of REACH lists a series of broad categories of substances for which registration is deemed inappropriate or unnecessary. Substances included in Annex V are also exempted from the supply chain communication and downstream user provisions, as well as evaluation.
The registration exemption applies to the substances as such, provided however that they meet the conditions for the exemption which are given in the particular category of Annex V.
If you need more detailed information on any category of substances, you can find this in the Guidance for Annex V, which gives explanations and background information for applying the different exemptions and clarifies when an exemption can be applied and when not.
Where necessary, in the interests of defence, Member States may allow for exemptions from the REACH Regulation in specific cases for certain substances on their own, in a mixture or in an article (Article 2(3) of the REACH Regulation).
Furthermore, Member States are not obliged to supply information the disclosure of which they consider to be contrary to the essential interests of its security (Article 346 TFEU1).
More information on national exemptions in the interest of defence in individual Member States is available on the European Defence Agency website.
When a substance is used in a medicinal product within the scope of either:
- The Regulation on Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency ((EC) No 726/2004); or
- The Directive on the Community code relating to veterinary medicinal Products (2001/82/EC); or
- The Directive on the Community code for medicinal products for human use (2001/83/EC); the substance does not have to be registered under REACH for that use.
The exemption does not distinguish between active or non-active ingredients as it applies to any substance ‘used in medicinal products'. Excipients used in medicinal products are therefore also exempted from registration.
Amounts of the same substance used for other uses than pharmaceuticals are not exempted. Only the amounts of the substance used in medicinal products are exempted from the registration obligation.
For further information, see chapter 2.2.3.2 of the Guidance on registration:
Active substances manufactured or imported for use in biocidal products are regarded as registered for the use in that biocidal product in the following situations:
- The active substance has been approved in accordance with Regulation (EU) No 528/2012 ("BPR"), or
- The active substance is under assessment in the review programme of existing active substances established under Article 16(2) of Directive 98/8/EC and continued under Article 89 BPR.
The list of approved active substances is available from the ECHA website:
To check which active substances are in the review programme, please see Annex II, part I to Commission Delegated Regulation (EU) No 1062/2014, also available from the ECHA website:
An exemption from REACH registration also applies in the following cases:
- The active substance is manufactured/imported for use in a biocidal product which has a simplified authorisation (Article 27 BPR)
- The active substance is manufactured/imported for use in a biocidal product which has a provisional authorisation (Article 55 BPR)
- The active substance is manufactured/imported for use exclusively in a biocidal product which is the subject of experiments or tests for the purposes of scientific or product and process-orientated research and development (Article 56 BPR).
If you manufacture or import a substance for biocidal and non-biocidal uses, you need to register it for the quantities of the substance used in non-biocidal products.
Active substances manufactured or imported for use in plant protection products, in accordance with Regulation (EC) No 1107/2009 concerning the placing of plant protection products on the market, are regarded as registered under REACH (for that use) if the active substance is approved and included in Commission Implementing Regulation (EU) No 540/2011 (list of approved active substances), or where the application for approval of the active substance is deemed admissible in accordance with Article 9 of Regulation (EC) No 1107/2009.
Amounts of the same active substance used for other uses than in plant protection products are not regarded as being registered even if they are approved. Also, other substances such as co-formulants, synergists, safeners and adjuvants are not regarded as being registered.
The Commission maintains an electronic list of the approved (and non-approved) active substances which is available at the following link:
A recovered substance should be registered as soon as it is no longer considered waste (when it reaches the end of waste criteria).
For further information on the end of waste criteria, see the following page:
To benefit from this exemption, you need to document that the following conditions apply:
- The same substance must have been registered.
- The substance must be the same.
- The company that did the recovery must have the information required by REACH available (e.g. safety data sheet).
For further information, see chapter 2.2.3.5 ‘Recovered substance already registered' in the Guidance on registration:
Annex IV of REACH lists a number of substances for which sufficient information is available to consider them as causing minimum risk to human health and the environment because of their intrinsic property.
These substances are typically of natural origin and the list of exempted substances includes, for example, water and nitrogen. Substances included in Annex IV are exempted from the registration, supply chain communication and downstream user provisions as well as evaluation.
The registration exemption applies to the substance as such, not to a particular use.
For more information see Annex IV of the REACH Regulation.
ECHA validates and quality checks the IT algorithms but does not examine all individual findings one-by-one. The IT results for a selected substance are checked and validated by the Member State competent authorities (MSCAs) during the manual verification phase (manual screening).
ECHA releases the shortlist of substances of potential concern in early January and Member States have until mid-Februuary to select substances for manual screening.
The manual screening can take from a few days to several weeks, depending on the complexity of the case. The deadline for screening is end May but for larger and more complex groups, there is some flexibility. However, if MSCAs wish to place the substance on the draft CoRAP list for next year, it is important that the deadline of end May is respected.
For more information on the screening process and timelines, please see the Common Screening Approach document: https://echa.europa.eu/documents/10162/19126370/common_screening_approach_en.pdf
We recommend monitoring the ECHA dissemination on a regular basis to see if some further actions have been started on your substance: http://echa.europa.eu/information-on-chemicals
The manual screening can result in the following outcome options:
- Candidate for compliance check (CCH): standard information requirements are not fulfilled: if the outcome is CCH and the substance is considered a priority for ECHA, then the substance would appear in the list of substances potentially subject to CCH: http://echa.europa.eu/regulations/reach/evaluation/compliance-checks
- Candidate for substance evaluation (CoRAP substance): in October each year, the draft CoRAP is published on ECHA’s website: http://echa.europa.eu/ information-on-chemicals/evaluation/community-rolling-action-plan.
If the outcome of the manual screening is as a candidate for substance evaluation, then the substance would appear in the draft CoRAP. - Candidate for further regulatory risk management:
- Risk management option analysis (RMOA): if the outcome of the manual screening is RMOA, then the substance will appear on the Public Activities Coordination Tool (PACT) available at: http://echa.europa.eu/pact
- Proposal for harmonised classification and labelling (CLH) at EU level: if the outcome is a CLH proposal, then once the Member States are ready to prepare the dossier, the intention to do so will be available at: http://echa.europa.eu/registry-current-classification-and-labelling-intentions
- Need for further assessment before the SVHC properties are confirmed and further regulatory risk management can be decided (e.g. the substance needs to be further assessed and discussed by the PBT or ED expert groups): if the outcome is further assessment and the need is to investigate PBT/ED properties, then this information will be available on: http://echa.europa.eu/pact
- Need for other action (e.g. enforcement or action under other regulations);
- No need for further action.
If the outcome of the manual screening would be no action, the substance would not appear in any follow up action. However, this information is not made publicly available and is not communicated to registrants. Manual screening cannot be completely comprehensive and even though the MSCA considers that no action is needed at present, the substance may be selected for manual screening again at a later date. New information or new uses may change the outcome of the screening.
Please note that it is up to the Member States to decide on the outcome of the manual screening.
In general if there is enough information to conclude that there is a concern, then the immediate follow up would be RMOA or CLH. If there is a need first to generate additional information, then either compliance check or substance evaluation would be selected by Member States. Registrants can influence the outcome of manual screening by timely updating their dossiers with any additional information/clarification
MSCAs make their own selection of the substances from the shortlist and not all substances are necessarily selected. In the case of large groups of substances, two or more MSCAs can collaborate during the manual screening. We don’t publish the evaluating MSCA but whenever a regulatory activity is initiated on a substance, the Member State or authority initiating the activity is published on our website, along with their contact details. When a regulatory action is started on a substance, a face-to-face meeting might be possible if initiated by the MSCA but no such interaction is foreseen during manual screening.
We check to see whether there are any ongoing activities with the substance or similar substances before shortlisting. For individual substances, depending on the nature of the process and the concern examined in these ongoing processes, we may exclude them from the shortlist. If a substance with an ongoing RFEACH/CLP regulatory process belongs to a group of substances that merits to be manually screened together by the same Member State, that substance will appear as a group member. ECHA informs the evaluating MSCA of the ongoing process for that substance.
The shortlist for manual screening is only released to MSCAs once a year and aims to identify candidates for several REACH and CLP processes. Member State competent authorities perform the manual screening. Compliance check can be an outcome of this manual screening. However, substances may be selected for Complicance Check by other means.
The list of potential candidates for CCH is updated several times a year, published on ECHA’s website and aims to identify candidates with specific data gaps that can be addressed with CCH.
Yes, the CSR will be examined during the manual screening.
During manual screening, MSCAs determine whether regulatory action is needed for a substance and if so, which is the most appropriate action. It is therefore the first step for most substances entering the other processes under REACH or CLP. However, Member States can propose a substance for the CoRAP or initiate regulatory action on any substance at any time so it is possible for a substance to go straight to the CoRAP, RMOA (PACT listing) or regulatory risk management without manual screening. MSCAs may select substances based on their prioritisation schemes, although every effort is made so that all MSCAs and ECHA collaborate in common screening.
Note though that there is a general agreement among Member States to have a RMOA done first before proposing a substance for being identified as an SVHC or to go through the restriction process.
It is not up to the registrant to decide whether one substance should be deleted or not from the shortlist. For a substance considered as not being hazardous and/or for which there is no need to go for further action based on uses and exposure information, the outcome of the manual screening done by the Member States will most probably be no need for further action for the time being.
During IT screening, ECHA invites Member State Competent Authorities to propose substances meeting their national priorities to be added on the shortlist. Such substances can be identified based on many issues but are usually not found by IT screening as being substances of potential concern.
An "ECHA-term" page is available from the front page of ECHA’s website. Acronyms mentioned during the webinar presentation may not all be found there. We recommend you first to consult the document entitled "a common screening approach for REACH and CLP processes".
The document can be found at: https://echa.europa.eu/documents/10162/19126370/common_screening_approach_en.pdf
They are of very different nature. The manual screening process aims at identifying substances of potential concern for regulatory action. The manual verification done in conjunction with the automated completeness check aims at verifying certain elements of the registration dossier that cannot be checked automatically, to ascertain that all the information required by the legislation has been included in a REACH registration. Further explanation of manual verification at completeness check is given here: https://echa.europa.eu/documents/10162/13652/manual_completeness_check_en.pdf
There is in principle no differentiation made between the different substance types (well-defined vs UVCB). Our screening algorithms are applied to registered substances regardless of the substance type (mono-constituent, multi-constituent or UVCB). A potential concern can be identified based on their individual constituents. It is important that the compositional information of UVCBs is properly documented in the REACH registrations.
A substance is not excluded from screening just because it has been evaluated under the old ESR process but any conclusions reached during this evaluation should be taken into account during manual verification. ECHA is using a range of internal and external regulatory sources to ensure that previous or on going actions on the same or similar substances are identified and are made available to the MSCAs so that their relevance for the newly identified concerns are fully addressed.
A substance is not excluded from screening just because it has been evaluated under the old ESR process. but aAny conclusions reached during this evaluation should however be taken into account during manual verificationscreening. ECHA is using a range of internal and external regulatory sources to ensure that previous or on going actions on the same or similar substances are identified and are made available to the MSCAs so that their relevance for the newly identified concerns are fully addressed.
If a REACH registration is ceased, the related registration dossier is not taken into consideration for creating the shortlisting. However, if the cease of manufacture occurs after the shortlisting the possible follow-up processes will consider the new status of the registration.
The European Commission announced last June scientific criteria to determine what is an endocrine disruptor under the pesticides and biocides regulations. Although there are no criteria for identifying endocrine disruptors under REACH, at the moment they are identified case-by-case based on the WHO/IPCS definition.
According to the recommendation of the EU Commission's Endocrine Disruptor Expert Advisory Group (ED EAG), factors such as severity, irreversibility, lead toxicity and potency can be considered to characterise the hazard potential of an endocrine-disrupting substance, e.g. when assessing the relevance of a substance for consideration in regulatory terms.
Therefore, on the basis of the WHO/IPCS definition, it is possible to search for indications that a substance may be endocrine active and/or elicit adverse effects that are (potentially) mediated by endocrine modes of action and the lack of criteria under REACH is not a reason to postpone screening activities.
For the purposes of screening, it is sufficient if algorithms identify endocrine active substances although some scenarios provide suspicion of adverse effects potentially caused due to an ED mode of action. The substances will then be manually screened and any ED activity and/or adverse effects will be scrutinised.
Please refer to the screening definition document for more details on our current approach in identifying potential ED substances in common screening algorithms.
Endocrine disruption may manifest itself in toxicological and ecotoxicological standard (in REACH Annexes) tests. As explained above, and in more detail in our definition document, common screening is looking for such evidence. Therefore, if there is a concern for ED properties based on the dossier, or external data, registrants might perform or propose further specialised in vitro or in vivo ED tests as necessary. In case such test results are available they should be included in the registration dossier, even if they do not constitute standard information requirements.
The OECD Conceptual Framework for Testing and Assessment of Endocrine Disrupters lists the OECD Test Guidelines and standardised test methods available, under development or proposed that can be used to evaluate substances for endocrine disruption and could provide a guide to the tests for endocrine disrupters’ assessment.
For the IT-screening, we use external lists of suspected ED such as the Commission, WHO, TEDX and SIN lists. Further evaluation steps during the manual-screening conducted by the MSCAs will assess the reliability of the available evidence on a case by case basis. However, the listing of a substance in an “ED-list” is never used as stand-alone criterion to suspect a substance as potential ED but always in combination with other information. The different sources of information and how these are used are described in the definition document that is updated annually at: http://echa.europa.eu/screening.
Common screening uses a wide range of QSAR models to detect likely endocrine disrupting properties. Moreover, substances confirmed to be endocrine disruptors are used as “seeds” to identify structurally similar substance that may exert similar properties. Finally, common screening uses external (external means data outside IUCLID registration dossiers) databases with confirmed/suggested endocrine disruptors and external databases with in vivo and in vitro data related to endocrine disruption.
When substances are grouped because there is evidence that similar hazards are to be expected, then the datasets of all substances are considered together in manual screening. This approach may, to some extent, alleviate issues with information deficiencies for one or more of the substances in the group that in principle could not be assessed on their own without generating further information. However, if there are reliable experimental data on the same substance and these prove that the suspectd concern does not hold then these data will overrule any conflicting evidence from similar substances (and even question the robustness of the grouping).
The grouping algorithms consider all information in registration dossiers, including migrated read-across endpoint study records. However, the clarity of such information is significantly reduced compared with a dossier that has been submitted to benefit from the additional features provided by IUCLID 6. This version of IUCLID includes a much clearer presentation of read-across information using two separate endpoint study records for the source and target substances. Moreover, the automatic migration may have introduced artefacts in the test material identifiers due to initially poorly submitted test material information in IUCLID 5. In such case the grouping algorithm may be less accurate. In the interest of efficiency, registrants are advised to consider updating their registration dossiers so that they can manually examine the correctness of the migration and use the new features of IUCLID 6
Some entries in the Candidate List of SVHCs are indeed group entries covering several substances. To find out the substances covered by such entries, please consult first the support document which defines in more details the substances belonging to the entry. The document is accessible by clicking the last column of the relevant Candidate List entry. ECHA also normally publishes a non-hexaustive list of substance identifiers on the same webpage in a separate document entitled “Non-exhaustive list of Substance identifiers”.
For substances shortlisted as part of a group, the group members were included in the letter sent to registrants (as separate annex). If no group was indicated, your substance was shortlisted as an individual substance.
If a substance is already included in a group it will be manually screened by the Member States. Registrants cannot influence grouping based on structural similarity. However, they may be able to influence grouping based on read-across or categories by strengthening the read-across argumentation to explain the similarities or dissimilarities between substances. Read-across approaches can also be removed and testing may be proposed instead to fulfil the information requirements.
No this is not made available to registrants, however in the letter sent to registrants information on shortlist substances that are part of the same group is given. For structural similarities search the OECD QSAR toolbox can be used.
Yes, groups are based on read-across linkages for the most relevant endpoints even if the substances are structurally different. Of course during manual screening the validity of RA/structural differences will be further scrutinized.
It is a reasonable initial assumption that similar substances might show similar toxicological properties. The presence of similar functional groups should be picked up by our similarity assessment. Further scrutiny on how structural differences impact toxicological profile are made during the manual screening. Generally, screening uses quite tight structural similarity criteria.
No, groups formed based on algorithms are not fixed. In the first instance the whole group will be subject to manual screening as a whole. However, based on the conclusion of the manual screening it may be that the group is found invalid, or it needs to be split into smaller groups containing substances with likely similar (eco)toxicological modes of action. With regard to any detected information requirements that are not met, it is possible that one (or some) of the substances in the groups may be selected for testing, typically the substance expected to represent worst case.
You are not formally required to update your registration. However, if you consider that the information in your own registration is not up-to-date, or that you have additional data that may assist with the assessment of your substance, ECHA recommends updating your dossier by the deadline specified in the letter you received. If the information to be updated is submitted jointly, the lead registrant can update the registration they submitted on behalf of the co-registrants. The updated information will be taken into account during the manual screening carried out by the MSCAs or during any potential follow up regulatory action.
ECHA is aware of the short time between the receipt of the letters and the start of the manual screening work by the MSCAs
On a monthly basis, ECHA provides the MSCAs with a report with all the updates submitted for the shortlisted substances. Therefore, if a substance is selected for manual screening, MSCAs will be informed about dossier updates, in case these updates cannot be submitted before the start of the manual screening work.
Please note that dossier updates not considered during the manual screening will be valuable if the substance is selected for further regulatory processes as a result of this manual verification. Unfortunately, we cannot send the letters earlier than January because this is when the shortlist is finalised.
The aim of the letter campaign for shortlisted substances is to inform registrants about the shortlisting and to invite them to review the related registration dossiers and consider to update them in particular for information on uses and tonnage per use and possibly on the potential hazard properties, as indicated in the letters, before the manual screening starts.
However, if the registrants are confident that the information in the dossiers is up-to-date, then no action is needed. Registrants can also attach a clarification document/peer reviewed papers in an updated dossier if they wish so.
Registration updates are only relevant if new/revised information is to be included. If the information in your registration is correct, you do not need to update your dossier or respond to the letter you received.
If relevant new/revised information is to be included in a dossier submitted jointly, an update by the lead registrant is sufficient if this new/revised information is valid for all registrants. However, if the new/revised information to be included in a dossier updated is members’ specific (e.g. information on substance composition or on specific uses) then it could be that only certain members need to update their dossiers.
The deadline set in the letter for shortlisted substances is the date when MSCAs may start the manual screening. It is important to clarify that these are informative letters, so there is no legal obligation to update registration dossiers. However, it is in the registrant’s own interest to review their dossiers and ensure that information on potential hazard properties and uses is up-to-date.
One of the aims of the letter campaign and interaction with Registrants in the early phases of common screening is to increase transparency of authorities' work in the early stages of substance selection and to give Registrants the possibility to clarify the potential hazard and use profile of their substances. The improved quality of the registration dossiers will in turn provide authorities a more solid basis for deciding on the need for further actions.
Therefore, if we have in registration dossiers the right information on hazard and uses this will impact the way substances are selected for further processes. Which means, for example, that if a substance is mainly handled under rigorous containment the authorities do not want to spend time on screening it. It is in fact in the interest of both authorities and industry that we focus on substances that matter and for which uses are of relevance from a regulatory risk management perspective.
Registration updates are valuable also if received after the manual screening. In such case, the dossier update will be looked at if the substance is selected for further regulatory processes as a result of this manual screening. Please note that ECHA provides the MSCAs with a report with all the updates submitted for the shortlisted substances on a monthly basis.
The detailed description of the uses and exposure related screening criteria used to prioritise substances is available in the screening definition document updated annually at: http://echa.europa.eu/screening. Priority is given to those substances having a high tonnage in uses considered as being wide dispersive.
Note that IUCLID6 has been further enhanced from a use and exposure perspective also taking into account the needs from the screening by authorities. Registration information is the main source of information for screening and it is therefore important that this information is up to date. We are also using external sources of information as listed in the screening definition document to support registration information.
The aim of the screening is to prioritise those substances that matter and for which the need for regulatory action is expected in order to best use the resources of both authorities and industry. For instance a substance registered at 100 tonnes per year for which 99 tonnes goes to intermediate uses and only 1 tonne to wide dispersive uses would be of lower priority compared to a substance with the same tonnage but for which 99 tonnes would go to wide dispersive uses.
This highlights the fact that the following information are needed to best assign priorities to substances:
- Information on the scope of the regulatory status of the uses (e.g. intermediate use, biocide);
- Tonnage per use;
- Whether the use is under rigorous containment to decide whether the substance has wide dispersive use or not.
Authorities are interested in substances where the uses can be regulated by REACH. For each REACH process, exemptions are available and documented both in the REACH Regulation but also in the screening definition document. It is also important to note that this information is used to set priorities among substances.
The screening has so far considered those substances with wide dispersive uses (i.e. professional, consumer and/or article service with potential for exposure to human or release to the environment) as a high priority. However, industrial uses can also be considered wide dispersive and may be prioritised later on as described in the screening definition document.
If professional, consumer uses and article service life are considered by default as being widespread (many users and/or many sites) this is not the case for industrial uses for which it is up to each registrant to indicate whether they believe their uses only take place for instance at few sites and to justify it in IUCLID6.
Substances that have no widespread uses are considered of lower priority for the time being and will not be included in the short-list for manual screening by Member States except if they are indications that such substance can be used as a substitute for a structurally similar substances already under regulatory scrutiny.
It is important to ensure that the uses reported in your registration dossiers are up-to-date and reflect uses that are on the market.
The screening is done at the level of the substance and therefore considers information on uses coming from all registration dossiers for that substance.
As a consequence, it is in the interest of all registrants to have up to date uses reflected in the registration dossier.
We are aware that it may be difficult to gather information on uses and tonnages per uses from downstream users in the supply chain. However, it should be clear to all that if some uses are relevant for further regulatory action, the Member State will consider the substance for further action even though it may only be present in one member dossier.
It may be difficult and time consuming at the level of the manual screening to gather such information however if the substance is moved to further action then there will be more time and more possibilities for exchange with the Member States and this information can still influence the outcome of the action envisaged for that substance.
If it is difficult to communicate with your downstream users, we would encourage you to try to explain those considerations. For confidentiality, it will potentially be easier for the downstream user to provide such information directly to the Member State in charge, for instance, of substance evaluation or RMOA. However, it should be kept in mind that providing clarification on the uses as early as possible is in the interest of registrants, downstream users and authorities.
In addition, in ECHA's view downstream users would not disclose confidential business information (CBI) if they break down the tonnage received from their supplier into generic type of use (possibly making reference to the cumulative tonnage fields in IUCLID 6:
- consumer uses;
- widespread professional workers;
- industrial use;
- service life.
Both lead registrants and co-registrants are responsible for updating the information in their registration dossiers.
The primary input to the common screening algorithms come from REACH registration dossiers. With regard to submitted data, common screening also uses the data in the C&L inventory, although more weight is usually given to classifications that are underpinned by test data in the registration dossiers. In addition to the submitted data in REACH dossiers and C&L notifications, the algorithms use external tools and databases to derive molecular structures from the chemical names and numerical identifiers, such as the CAS number and the EC number.
These structures are used to run predictions for fate and (eco)toxicological properties using models. Finally, the algorithms use external lists, such as the SIN list: (http://chemsec.org/what-we-do/sin-list), and the assessments from other regulatory regimes, such as the IMAP programme: (http://www.nicnas.gov.au/chemical-information/imap-assessments/imap-assessments) in Australia. ECHA is also using datasets used for QSAR model development in case the screened substance is identical or similar to one of the tested substances in the training sets.
The different sources of information and how these are used are described in the definition document that is updated annually at: https://echa.europa.eu/screening.
The external sources used are carefully selected based on the available information, which includes discussion with the data providers, e.g. other regulatory authorities and non-governmental organisations. Nevertheless, ECHA is not in position to assess every individual entry in the external lists.
ECHA provides all algorithm results to Member States that enables the Member State experts to verify the relevance of all findings before deciding on any required regulatory actions.
If ECHA possesses additional information, such as toxicological assessment accompanying non-governmental substance priority lists, this information is provided to Member States.
ECHA is making every effort to provide as much information as possible in the communication letters. However, due to the high number of letters and complexity, ECHA is not in the position to list all technical information that led to shortlisting in the communication letters, such as the entry in the particular external list that was matched against a constituent, impurity or additive in the registration dossier.
The external sources are listed in the definition document, which can be consulted for more information. It is available at: http://echa.europa.eu/screening.
The C&L Inventory is one of the sources used to identify substances of potential concern as it represents an overview of how the substance is classified on the EU market. We take into account the number of notifiers behind each classification and the consistency of the notifications. Diverging classifications in the C&L Inventory can reveal disagreements among manufacturers and importers on how to classify the substance, which might require regulatory action such as harmonised classification and labelling. The IT screening findings are checked by MSCAs during manual screening. In general, we place a higher emphasis on classifications coming from REACH registrations but those coming from notifiers often provide valuable insights into the substance in question.
ECHA makes every effort to include as much information as possible in the letters.
However, due to the high number of letters and complexity, ECHA is not in the position to list all technical information that led to shortlisting in the communication letters, such as the entry in the particular external list that was matched against a constituent, impurity or additive in the registration dossier.
Further details are provided in the definition document updated annually at: http://echa.europa.eu/screening.
It is advisable to consult this document if you have received a letter pointing to an external source or QSARs.
We use a variety of commercial and public predictive tools, such as the public tools OECD QSAR Toolbox (http://www.qsartoolbox.org/) and EPISUITE (http://www.epa.gov/tsca-screening-tools/epi-suitetm-estimation-program-interface). The tools are of various kinds, including classical QSAR models and predictive methods based on structural alerts.
ECHA is not in a position to publish the full set of tools used for screening, especially if they are commercial. We try to cover all major REACH endpoints, often with more than one tool, so that the tools can be used in combination to assess the consistency of the predictions.
For certain endpoints, the model predictions are only used as supporting information to other hazard evidence, such as the listing of the substance in priority lists of non-governmental organisations.
The algorithms use applicability domain considerations when appropriate. Moreover, multiple models are often used in combination to be able to check their consistency. For models where the expected error may be larger, predictions are only used as supporting information to other findings, such as weak/inconclusive evidence of hazard in experimental studies reported in the registration dossiers. The validity and relevance of all findings, including the model predictions are assessed by Member States during manual screening.
ECHA is independently running a suite of predictive tools but is also using the model results found in registration dossiers. The screening algorithms use all endpoint study records in the registration dossiers, but they give different weights depending on the reliability of the information provided. This is to ensure that good quality experimental studies have more weight compared with predicted results, although contradicting results are also identified for further investigation.
Common screening uses an extended set of tools to generate molecular structures from the identifiers provided in registration dossiers and C&L notifications. The algorithms consider all registrations/notifications, all compositions and all reference substances in ECHA’s databases.
For each reference substance, we use all identifiers provided, such as the CAS number, CAS name, IUPAC name, SMILES, InChI and synonyms. In this way, we can ensure that the algorithms are run on the broadest possible set of chemical structures associated with the registered substance.
This means that the algorithms also generate predictions for minor constituents, impurities and additives, but their concentration and frequency of occurrence in the joint submission are used as criteria for shortlisting.
During manual screening, Member States examine the validity of the predictions, which on some occasions may be compromised due to the generation of erroneous structural information, e.g. when a registrant provides a synonym that points to a molecular structure other than the constituent, impurity or additive indented to be included in the registration.
This information is needed to verify the composition of your substance and to ensure that the chemical identifiers, such as IUPAC name or CAS number, are appropriate.
ECHA requires, as a minimum, ultra-violet (UV), infra-red (IR) and nuclear magnetic resonance (NMR) spectra (Annex VI section 2.3.5, REACH). You can also provide a mass spectrum in place of an NMR spectrum. For some substances, this information is not sufficient or appropriate and, in such cases, you need to provide other types of spectral data. For example, in the case of inorganic substances, X-ray diffraction (XRD), X-ray fluorescence (XRF) or atomic absorption spectroscopy (AAS) are likely to be more appropriate techniques.
No. ECHA protects the confidentiality of the analytical information submitted by (potential) registrants.
An acceptable justification should be based on technical feasibility or scientific necessity, meaning robust scientific argumentation.
Yes. The sum of the typical concentrations of each constituent should add up to 100 %.
When preparing your dossier to be submitted to ECHA, we recommend that you also read the following documents:
- Guidance for identification and naming of substances under REACH and CLP: http://echa.europa.eu/documents/10162/13643/substance_id_en.pdf
- The relevant Submission Manuals at: http://echa.europa.eu/manuals
- Question and answers on Substance Identification http://echa.europa.eu/support/qas-support/qas
- Question and answers on Inquiry http://echa.europa.eu/support/qas-support/qas
Yes. Ionic substances in an aqueous solution are exempted from registration only if:
- All starting substances (salts, acids and bases) of the aqueous solution are registered;
- None of the salts in the aqueous solution is isolated from the solution; and
- The salts remain in their ionic form in the solution.
In this particular case, the third condition is not fulfilled, since the salts do not remain in their ionic form in the complex. Therefore, this exemption does not apply and the complex would be subject to registration. This case is discussed in Attachment 1 ‘Ionic Mixtures' of the Guidance document to Annex V.
Complexes consisting of chelated ions must be registered if they are themselves manufactured, imported or placed on the market. However, there are different exemptions in Annex V which could be considered, for example if the complex results from a chelating agent functioning as intended (‘substances which are not themselves manufactured, imported or placed on the market and which result from a chemical reaction that occurs when: (a) a [...] chelating agent [...] functions as intended').
For the purpose of registration, hydrates of a substance and the anhydrous substance are regarded as the same. If the anhydrous substance is registered by the manufacturer/ importer, and if a company uses that substance to manufacture the hydrated substance, this company is regarded as a downstream user. Therefore, the respective registration dossier must include the necessary information for the anhydrous and the hydrated substance occurring within the supply chain, i.e. the chemical safety assessment with exposure assessments and risk characterisations of hydrated substances and the water-free substance. This information needs to be communicated along the supply chain in safety data sheets which should include all the necessary facts, e.g. exposure scenarios and identified uses for the water-free as well as the hydrated substance which occur within the supply chain. The registration number of the registered anhydrous substance must be specified in section 1 of the SDS. The provisions foreseen for downstream users according to Articles 37 and 38 can be implemented for both the hydrates of a substance and the anhydrous substance.
Registrants need to take into account in their CSA the potential implications of specific forms of the substance on downstream uses, in particular when the form is changed during downstream use.
Yes. To confirm the presence of the ions, you need to provide analytical data for the identification and quantification of each ion.
Provided your well-defined substance consists of all possible stereo isomeric forms as the main constituents of that substance, you can identify your substance in IUCLID section 1.1 using only the IUPAC name of the substance without specifying the stereochemistry. However you should still identify your substance as a multi-constituent substance. Information on the identity and concentration of each of the individual stereoisomers should be reported in IUCLID section 1.2.
When your substance contains impurities that you are unable to identify, you need to create a generic reference substance for them in IUCLID. In the reference substance, you need to state the following:
- "unknown impurities" in the IUPAC name field; and
- the number and the individual concentration range of each unknown impurity in the remarks field.
In addition the typical concentration and concentration range of the unknown impurities has to be provided.
An illustrative example on how to report unknown impurities in IUCLID can be found in section 9.4.2 of the manual How to prepare registration and PPORD dossiers, which is available at: http://echa.europa.eu/manuals
Yes. You need to report concentration ranges (i.e. both minimum and maximum values) for each constituent. These should be representative of the substance as manufactured/imported and you can take them from, for example, certified specification limits that often form part of a certificate of analysis (CoA). It is important that the concentration ranges are realistic and do not cover different substances.
REACH-IT will provide you with a list number once your registration passes the business rules. You will be able to download this automatically created EC entry as an i6z file from REACH-IT. You will be requested to assign this list number when submitting any update of your registration.
You must report the chemical name of your UVCB substance in the ‘IUPAC name' field of IUCLID Section 1.1. You can find the naming conventions for deriving the chemical name of UVCB substances in Chapter 4.3 of the Guidance for identification and naming of substances under REACH and CLP. An example of a chemical name for UVCB substances is "Oligomerisation reaction products of formaldehyde and phenol".
You must report the chemical name of your multi-constituent substance in the ‘IUPAC name' field of IUCLID Section 1.1. The generic format for the chemical name is "Reaction mass of" followed by the name of each main constituent separated by "and". In principle, the name of the main constituents should follow the IUPAC rules. An example of a chemical name for multi-constituent substances is "Reaction mass of ethylbenzene and m-xylene and o-xylene".
You should normally report each individual isomer and their typical, upper and lower concentration levels separately in IUCLID Section 1.2.
As a deviation, you may report the isomers under one entry in IUCLID Section 1.2. This approach may be considered appropriate when the isomers are present as a racemic mixture or when the number of isomers is large. In that situation, you should do the following:
- Specify the overall typical, upper and lower concentration level of isomers covered by the entry in the relevant IUCLID Section 1.2 fields;
- Indicate the relative ratio of isomers in the "Remarks" field in the repeatable block for that entry. If the relative ratio of isomers varies, you will need to report it in the form of a range;
- Indicate that specific isomers have not been reported separately in the "Justification for deviations" field.
No. The "Legal entity composition of the substance" should only be used to report compositions of the substance on its own that you manufacture or import. For instance, if your substance is formulated with a solvent upon manufacturing, you should only report the composition of the substance without the solvent as "Legal entity composition of the substance".
If your substance is only made available as a component of a mixture, the "legal entity composition of the substance" to be reported should still be for the substance on its own.
You should not confuse a mixture with a multi-constituent substance or a UVCB substance. Under REACH and CLP, a mixture is obtained by blending two or more substances without a chemical reaction. Substances within a mixture should be registered separately. A multi-constituent substance or a UVCB substance is normally the result of a chemical reaction. A multi-constituent substance or a UVCB substance should be registered as such.
You should proceed as follows:
- Assign the reference substance for the anhydrous substance and select the corresponding type of substance (mono-constituent, multi-constituent or UVCB) in Section 1.1.
- For technical reasons, report the composition of the anhydrous substance as the first composition with the composition type "Legal entity composition of the substance" in Section 1.2.
- If you manufacture or import the anhydrous substance, you should report the composition of that substance as the first composition.
- If you do not manufacture or import the anhydrous substance, you will still need to report a composition corresponding to the anhydrous substance as the first composition in Section 1.2. In this case, we recommend you to report a theoretical composition where the reference substance for the anhydrous substance is reported at a typical concentration of 100 % (w/w). Select "Legal entity composition of the substance" as the type of composition. In the "Description" field, indicate that the reported composition is theoretical. In the "Justification for deviation" field, indicate the following: "This composition is neither manufactured nor imported. It is only reported for technical reasons because the derogation for the registration of hydrates is applied".
- Report all other relevant compositions, including the composition of the different hydrates that are covered by the registration. For the compositions referring to hydrates, indicate "Hydrate covered by the registration of the anhydrous substance" in the "Justification for deviation" field. The ‘Type of composition' of the reported hydrate compositions should be ‘legal entity composition of the substance'.
You should provide sufficient analytical information to verify the identity of the monomer you register. The analyses typically consist of spectral and chromatographic data and a chromatogram of the original monomer or other substance used in the manufacture of the polymer. For further information, please consult Q&A 72 agreed with national helpdesks.
No. You should provide sufficient information in IUCLID Section 1.4 to identify the monomer to be registered.
If you encounter difficulties in getting hold of the information from the non-EEA manufacturer/supplier of the polymer, you may consider one of the following options:
- Propose to the non-EEA manufacturer of the polymer that they appoint an only representative (OR) in accordance with Article 8 of the REACH Regulation;
- Identify the non-EEA manufacturer of the monomer and request the analytical information directly from them;
- If an OR has already been appointed by the non-EU manufacturer, propose to that OR to take on the legal responsibility envisaged in Article 8 of REACH for the polymer importers (see also Q&A 834).
You may also consider any relevant scientific method to fulfil this information requirement. Should you consider information that is not limited to analytical data on the original monomer, we recommend you follow a clear and transparent approach carefully documented when reporting the information in IUCLID Section 1.4 of your registration.
The substance is in principle identified as an inorganic UVCB due to the variability of the molecular formula.
In this case please select “inorganic” as origin under the Type of substance heading of IUCLID section 1.1.
The variability in the stoichiometry should be reported under the molecular formula field (e.g. MxGyO2, x=a-b, y=c-d).
In case only one constituent block is reported in section 1.2 of the IUCLID dossier, a justification for reporting a single constituent in the composition of a UVCB substance (expected to have a variable complex composition) should be provided in the “justification for deviations” field.
One example of a valid justification would be:
“The substance is a UVCB due to variations at the elemental level and these variations cannot therefore be represented by reporting different constituents but only in the form of a variable molecular formula.”
Yes. The manufacturing process description is one of the identifiers for UVCB substances. The information should be included in the "Description" field of IUCLID Section 1.2. The description generally consists of the following:
- Identity and ratio of starting materials;
- Description of the relevant manufacturing steps in the order they occur;
- The relevant operating parameters applied to control the composition (e.g. temperature, pressure, solvent, catalysis type…);
- Details on any extraction/isolation/purification step.
In addition, you can also report the reaction schemes or process workflows to complement the description of the manufacturing process as an attachment under the "Attached description" heading of IUCLID Section 1.2.
No. The chemical name alone does not include all the process circumstances determining the composition of the UVCB substance and therefore its identity. For further details on the information expected to be reported on the manufacturing process of UVCB substances, please consult the Q&A 1199.
You first need to define which information on the manufacturing process is relevant for the identification of the UVCB substance. The Q&A 1199 will assist you in deciding which information on the manufacturing process matters for the identification of your UVCB substance.
The more you know about the composition of the UVCB substance, the less you will be dependent on the manufacturing process to identify your substance. For instance, you may not need to specify the ratio of reactants used to manufacture the substance if you can define the reaction yield and the content of residual starting materials from the compositional information. Be aware that in this case you will still need to explain why some elements of the manufacturing process expected to be submitted are not provided, as indicated in Q&A 1318.
If the missing information on the manufacturing process prevents you from identifying the substance and the non-EEA manufacturer does not share this information directly with you, you may propose to the non-EEA manufacturer of the UVCB substance that they appoint an only representative (OR) in accordance with Article 8 of the REACH Regulation.
ECHA has also prepared a template that can be used to collect the necessary information from the non-EEA manufacturer. The template is available through our website here.
You need to provide all the relevant information on the manufacturing process in IUCLID section 1.2 as instructed in the Q&A 1199. If you consider that some of the information is not relevant, you need to clearly explain why this information is not included in the same field where the manufacturing process description is provided (i.e. in the ‘Description’ field of IUCLID Section 1.2).
No. The manufacturing process description is necessary to circumvent the limitations of identifying UVCB substances by their composition only. You therefore need to provide both types of information (composition and manufacturing process description) in IUCLID section 1.2. For further information on how to report the manufacturing process description and the composition of UVCB substance, please refer the Q&A 1199 and chapter 9.4.2 of the manual ‘How to prepare registration and PPORD dossiers’ available on the ECHA website: https://echa.europa.eu/manuals.
The manufacturing process description is one of the identifiers for petroleum UVCB substances. This description should contain the following information:
- The source material from which the petroleum stream is obtained. This could be crude oil in the case of straight-run atmospheric distillates and atmospheric residue. For other petroleum substances, the source materials are often intermediate stocks and the name of the petroleum stream(s) to which the refinery process is applied should be specified. For example, for reformate fractions the source material could be heavy naphtha fraction from the atmospheric distillation of crude oil; for vacuum distillates the source material could be the residue from the atmospheric distillation of crude oil; the source material for base oils is normally a vacuum distillate; the source used to manufacture a naphtha fraction as a result of cracking process could be a vacuum distillate or heavy atmospheric gas oil. For imported gas oil, the source material could be various petroleum streams derived from crude oil, however they need to be specified as far as possible.
- The refinery process(es) applied to the source material(s). This may include blending processes (blending to specification), for which specification described by an international standard (e.g. EN228, EN590, etc.) should be provided. For a light reformate fraction, for example, the refinery processes include crude oil fractionation, naphtha hydrotreatment, catalytic reforming and reformate fractionation.
- The description as to how the final fraction is collected (atmospheric distillation, distillate/residue fraction; vacuum distillation, distillate/residue fraction; solvent extraction, extract/raffinate phase, etc.).
- Specification of the registered substance. Measured typical values of boiling point range and carbon number range need to be provided. If a stream is defined in its EC description by other parameters, e.g. viscosity, they also need to be provided. The refinery specific process parameters (e.g. refining temperature, catalysts, solvents, etc.) do not need to be provided.
It is important to mention that all the relevant information on manufacturing process description should be included in the “Description” field in section 1.2 of IUCLID. Information on manufacturing process that cannot be provided as free text (graphs, schemes, etc.) should be provided in a document and attached as instructed in Q&A 1199.
REACH applies both to substances occurring in nature, as defined by Article 3(39) of REACH, and to their synthetic analogues.
However, Annex V to REACH states that the following substances occurring in nature are exempted from registration if they are not chemically modified: minerals, ores, ore concentrates, raw and processed natural gas, crude oil and coal. These substances can only be processed by certain means (e.g. dissolution in water, flotation), which are specified in Article 3(39) of REACH and do not include chemical modification (Article 3(40)).
Other substances occurring in nature are also exempted from registration if they are not chemically modified, unless:
- they meet the criteria for classification as dangerous according to the CLP Regulation (Regulation 1272/2008), or
- they are persistent, bioaccumulative and toxic or very persistent and very bioaccumulative in accordance with the criteria set out in Annex XIII, or
- they were identified in accordance with Article 59(1) at least two years previously as substances giving rise to an equivalent level of concern as set out in Article 57(f).
Further explanations and background information on the different exemptions in Annex V are included in section 2.2.3.4 - 'Substances covered by Annex V of the REACH Regulation' of the Guidance on registration: http://www.echa.europa.eu/guidance-documents/guidance-on-reach
The REACH Registration Q&As provide guidance on substances occurring in nature that are obtained by extraction processes.
For particular guidance on polymer substances occurring in nature, see section 3.2.1.3 - 'Case of a natural polymer or a chemically modified natural polymer' of the Guidance for monomers and polymers: http://www.echa.europa.eu/guidance-documents/guidance-on-reach
Since the synthetic analogues of naturally occurring substances do not meet the criteria for substances occurring in nature as defined in Article 3(39) of REACH, any manufacturer or importer of these substances in quantities of one tonne or more per year is required to register them.
Substances listed in Annex IV to REACH are exempt from registration. Modified substances derived from these substances are also exempt if the modified substance is still covered by the same EINECS entry; whether the same EINECS entry applies is a case-by-case decision. For example, for plant oils such as soybean oil (EINECS no 232-274-4; CAS no 8001-22-7) the physically modified derivatives are explicitly covered in the EINECS entry. Whereas chemical modification (e.g. hydrogenation) is not mentioned and hence considered not to be covered. For further information, see Article 3(40) of REACH and Section 2.2.3.3- 'Substances included in Annex IV of the REACH Regulation' of the Guidance on registration: http://echa.europa.eu/guidance-documents/guidance-on-reach
In a surface-treatment process, the following substances are relevant:
- “Basis substance”: Substance in the form of particles on which the surface treatment is applied.
- “Surface-treating agent”: Substance reacting with the surface of the basis substance, i.e. the substance used as the starting material in the surface-treatment process.
- “Surface-treated substance”: Substance resulting from treatment of the basis substance with the surface-treating agent.
When a substance is surface-treated, the resulting surface-treated substance must not be registered as such under REACH. There are, nevertheless, consequences on the registration requirements for the basis substance and the surface-treating agent. These consequences depend on the form of the basis substance which is surface treated.
The basis substance is a nanoform
In case the form of the basis substance is a nanoform, the surface treatment is a characterisation parameter of the nanoform according to Annex VI to REACH. Any surface treatment of the basis substance and any change in the reaction conditions or in the molar ratio of the surface-treating agent generates a different nanoform that must be specifically reported in the registration dossier. Each nanoform of the basis substance must be characterised and specifically reported in the registration dossier according to sections 2.4.2 to 2.4.5 of Annex VI to REACH.
Each surface-treated nanoform covered in a registration dossier must be characterised according to Section 2.4.3 of Annex VI to REACH. This characterisation must include a “Description of surface functionalisation or treatment and identification of each agent including IUPAC name and CAS or EC number”. The registration dossier must also contain a specific set of hazard data (i.e. information requirements of Annexes VII to X) on the surface-treated nanoform. This set of hazard data must be clearly linked to the corresponding surface-treated nanoform characterised in accordance with Annex VI.
For guidelines on reporting nanoforms in a registration, see Q&As on nanoforms of substances and ‘Appendix for nanoforms applicable to the Guidance on Registration and Substance Identification’.
The basis substance is not a nanoform
In case the form of the basis substance is not a nanoform, any surface treatment of the substance would result in a “two dimensional” modification of particles, i.e. a chemical reaction only at the surface of the particles with the surface-treating agent. Therefore, a surface-treated substance cannot be regarded as a mixture nor be defined as a different substance based on the criteria of the Guidance for identification and naming of substances under REACH.
Concerning the basis substance:
A manufacturer or importer of the basis substance who applies the surface treatment, and an importer of the surface-treated substance must include the following information in the registration dossier of the basis substance:
- In Section 1.2 of the registration dossier:
- the identification of the basis substance; and
- the identification of the surface-treating agent and a description of the surface-treatment process (in the ‘Description’ field).
- Information on any hazards or risks specific to the surface-treated substance. These hazards or risks must be appropriately covered in the dossier by the classification and labelling and by the chemical safety assessment and resulting exposure scenarios.
Concerning the surface-treating agent:
The surface-treating agent must be registered by the manufacturer or importer of the surface-treated substance if they manufacture or import the surface-treating agent on its own in the EU/EEA. When the surface-treating agent must be registered, the registration dossier must include a description of the use “surface treatment” in Section 3.5 of the registration dossier.
Yes, biomethane obtained by the purification of biogas is considered to be covered by the exemption from registration according to Annex V entry 12 to REACH. This exemption does not refer to biomethane as such, but to biogas (consisting mainly of methane) produced by the biological breakdown of organic matter (e.g. agricultural waste, municipal waste, sewage) in the absence of oxygen. Nevertheless, biomethane obtained by the purification of biogas to remove undesirable components is still considered as biogas and is, therefore, exempted from registration according to Annex V entry 12 to REACH.
For methane processed from raw natural gas, the exemption in Annex V entry 7 to REACH applies. Methane obtained from other sources than fossils is not regarded as natural gas and is, therefore, not covered by this entry.
When brought in contact with water, chromium trioxide (EC number 215-607-8) forms two acids and several oligomers: Chromic acid (EC number 231-801-5), Dichromic acid (EC number 236-881-5), Oligomers of chromic acid and dichromic acid.
These chemical species are identified as substances of very high concern (SVHC) and included in the Candidate List1 as two separate entries.
Chromic acids and their oligomers generated in water from chromium trioxide are commonly referred to as an aqueous solution of chromium trioxide. With regard to the obligation to register, it may be justifiable in some specific situations described in the table below, to consider for practical reasons chromic acids and their oligomers as an aqueous solution of chromium trioxide. Hence, in these specific cases, chromic acids and their oligomers present in an aqueous solution of chromium trioxide can be covered by a registration dossier for chromium trioxide.
Important note: The presented approach is strictly limited to chromium trioxide and chromic acids and their oligomers generated from chromium trioxide in water. It derives from very specific aspects of the Chromium VI aqueous chemistry; the system in aqueous solution is a complex equilibrium between multiple chemical species which depends on several physico-chemical parameters and the different chemical species cannot be isolated from the aqueous solution. The approach can thus not be applied by analogy to any other substance.
Manufacturers and importers of chromium trioxide and chromic acids and their oligomers have to consider the following situations:
| Actor / Scenario | Legal requirement | Explanation |
|---|---|---|
| Manufacturer or importer of chromium trioxide who generates chromic acids and their oligomers in water | One registration according to Article 10 for chromium trioxide |
The generation of chromic acids and their oligomers by adding chromium trioxide to water and their further use have to be included in the registration dossier and have to be considered for the chemical safety assessment (CSA) and the chemical safety report (CSR). In case a downstream user (DU) generates chromic acids and their oligomers from chromium trioxide, this use has to be communicated up the supply chain and has to be included in the registration dossier. |
|
Importer of both chromium trioxide and chromic acids and their oligomers generated in water from chromium trioxide |
One registration according to Article 10 for chromium trioxide | The registrant has to register chromium trioxide and chromic acids and their oligomers in one dossier for chromium trioxide. It has to become clear from the registration dossier that chromic acids and their oligomers are also imported. Therefore, at least two compositions have to be provided in section 1.2 of the IUCLID dossier. The first composition refers to chromium trioxide; the second composition refers to the composition of chromic acids and their oligomers. A remark has to be entered to clarify the approach. The tonnage to be reported is the aggregated tonnage of both chromium trioxide and chromic acids and their oligomers. The tonnage has to be reported on the basis of equivalent chromium trioxide tonnage. |
| Importer of chromic acids and their oligomers generated in water from chromium trioxide | Registration according to Article 10 either for chromic acids and their oligomers generated from chromium trioxide or for chromium trioxide |
In case the importer decides to register chromic acids and their oligomers in a dossier for chromium trioxide, it has to become clear from the registration dossier that what is actual imported are chromic acids and their oligomers. Therefore at least two compositions have to be provided in section 1.2 of the IUCLID dossier. The first composition refers to the generic substance "chromium trioxide"; its purity and composition should be indicated as 100 %. The second composition shall refer to the actual composition of chromic acids and their oligomers which are imported. A remark has to be entered to clarify the approach. |
| Manufacturer or importer of chromic acids and their oligomers generated by alternative methods other than from adding chromium trioxide to water or Importer who is unaware of the manufacturing methods of the chromic acids and their oligomers |
Registration according to Article 10 for chromic acids and their oligomers | The approaches described above cannot be applied as the starting material for manufacturing chromic acids and their oligomers is not chromium trioxide or is not known. |
Please, also see Q&A=805 (Can an application for authorisation for chromium trioxide cover the use of the chromic acids and their oligomers generated from adding chromium trioxide to water?)
Yes. A request for a change of a substance identifier can be made by submitting a “Substance identity adaptation service web form”, which is accessible via ECHA’s website, at the following link: https://comments.echa.europa.eu/comments_cms/SubstanceIdentityAdaptation.aspx
It is technically not possible for registrants to change the substance identifier (EC or list number and name) of their registration dossier without requesting the change of identifiers service.
The request is normally made by the lead registrant, on behalf of the members of a joint submission. Member or individual registrants can also make the request. The request must include documentary evidence that all members of the joint submission agree with the changes specified in the request that affect their registration dossier.
Registrants will need to submit collectively the information included in the ‘Joint Submission Plan’ template, which is available on ECHA’s website: https://comments.echa.europa.eu/comments_cms/SubstanceIdentityAdaptation.aspx.
Once you submitted the request to change the substance identifier, the following steps take place:
- You receive an automatic email confirmation that includes the unique reference number of your request.
- ECHA assesses if the service can be provided based on the provided information. ECHA contacts you if the request is incomplete or cannot be provided.
- If ECHA considers that the service can be provided, it issues a service charge to each applicable registrant.
- After the registrants have paid the service charges, ECHA carries out the changes in REACH-IT.
- ECHA requests the registrants to update their registration dossiers using the new substance identifier and provides instructions on how to submit the dossier update.
- You and the applicable registrants submit dossier updates using the new substance identifier.
For further information about the service to change the substance identifier in a registration, please see “How to change your substance identifier” page on ECHA’s website, at the following link: https://echa.europa.eu/support/how-to-improve-your-dossier/how-to-change-your-substance-identifier.
The minimum charge is €300 (€600 for requests received from 1 November 2021) for each registrant requiring the correction of a substance identifier. More information is available on ECHA’s website: https://echa.europa.eu/support/how-to-improve-your-dossier/how-to-change-your-substance-identifier.
The REACH Regulation does not impose any timeframe on ECHA for processing a change of substance identifier request. ECHA aims to assess whether the service can be provided within 1 month. The whole change of substance identifier process normally takes between 3-6 months, if all required information has been provided, the request can be granted, the invoices are paid in a timely fashion, and the dossiers are successfully updated without delay by the registrants.
For more information on the SIP, please read appendix III - Substance identification and joint submission of data - of the Guidance for identification and naming of substances under REACH and CLP available on ECHA’s website at https://echa.europa.eu/guidance-documents/guidance-on-reach. You can also find technical instructions on reporting substance identity information in IUCLID 6 format in the manual “how to prepare PPORD and registration dossiers” available on the ECHA website.
The SIP is the collective agreement between companies. The boundary composition is the technical reporting to reflect the compositions of all members in the joint submission. In some cases, the boundary composition includes, besides the composition information, also any other relevant parameter(s), such as the manufacturing process.
Yes. ECHA will disseminate the information reported in the boundary composition record by extracting it from registration dossiers submitted in IUCLID 6 format and will publish it through our Information on Chemicals part of the ECHA website. Information on chemicals can be searched through the Search box on the homepage of the ECHA website.
The boundary composition record will not be published until the lead registrant updates the registration in IUCLID 6.
There is a Dissemination Preview tool included in IUCLID with which users can see what data will be published and how. We strongly encourage registrants to use this to check their dossier before submitting it to ECHA.
Where your composition is outside the boundary composition of an existing joint submission, the first step would be for you to contact the existing registrant(s) and discuss with him whether the Annex VII-XI data reported in the joint registration would be sufficient as to apply to your substance. Where it is agreed that the data is sufficient, it would then be necessary for the lead registrant to update the boundary composition record reported in the lead dossier to expand its scope. This would mean updating the boundary composition to cover the compositions of both your composition and that of the existing registrants.
All parties would need to ensure that the criteria applied are transparent and that the relevant Annex VII-XI data collected/generated demonstrably covers all agreed compositions.
No. You only have to include a boundary composition record the next time you update your submission.
However, we strongly encourage lead registrants to discuss the boundary composition with their members and potential registrants and update their dossiers as soon as possible.
For a new registration, it is now mandatory for lead registrants to include the boundary composition record when they submit their IUCLID dossier.
It is important that the compositions and parameters defining the boundaries of the substance covered by the joint submission are agreed by all the co-registrants and are clearly documented in the boundary composition. Accordingly, the boundary composition may need to be modified or extended following the request of any new potential registrant, if all co-registrants agree that part or all of the jointly-submitted data is also relevant for the substance manufactured or imported by this registrant. However, the lead registrant is not obliged to amend the existing boundary composition if he and the other co-registrants consider that by doing so they could no longer rely on the data to address the hazard properties of the extended composition. The rationale would need to be transparently documented.
More than one boundary composition record may be reported depending on how the co-registrants want to structure the reporting of their joint Annex VII-XI data. For example if there are compositions that have impurities/constituents that triggered PBT assessment and/or have different classification and labelling, these can be reported in their own boundary composition record in IUCLID.
Only the lead registrant, with the agreement of the other co-registrants, is required to report the boundary composition(s) in his dossier and keep it up-to-date. Any change made by the lead registrant at the level of the boundary composition will be visible to every member of the joint submission in REACH-IT.
The level of detail reported for the boundary composition is not dependent on whether the substance is hazardous or not. However, the level of detail for the boundary composition should be customised around the considerations used to construct the hazard dataset, including also the relevant considerations made at the impurity level to derive the classification and/or PBT assessment of the substance.
The Candidate List and additional useful information is published on ECHA's website at:
Substances fulfilling one or more of the criteria specified in Article 57 of the REACH Regulation can be identified as "substances of very high concern" (SVHC) and put on the "Candidate List". These SVHC can be:
- substances meeting the criteria for classification as carcinogenic, mutagenic or reprotoxic (CMR) category 1 A or 1B (in accordance with section 3.6 of Annex I to Regulation (EC) No 1272/2008)
- persistent, bioaccumulative and toxic (PBT) substances or very persistent and very bioaccumulative (vPvB) substances (according to the criteria of Annex XIII of the REACH Regulation)
- substances identified on a case-by-case basis for which there is scientific evidence of probable serious effects to human health or the environment which give rise to an equivalent level of concern (Article 57(f) of REACH Regulation), e.g. endocrine disruptors
The Candidate List is available on the website of ECHA. It has been established according to the procedure described in Article 59 of the REACH Regulation (SVHC identification).
The Candidate List is updated when substances are identified as Substances of Very High Concern. This is normally done twice per year (in June and December). To allow interested parties to be aware of substances which might be included in the Candidate List, a Registry of Intentions is published on the website of ECHA. As a producer, importer or supplier of articles, you are advised to regularly check the Registry of Intentions. This can help you to prepare for possible obligations that could arise when a substance is included in the Candidate List: https://echa.europa.eu/candidate-list-obligations.
You can find further information at: https://echa.europa.eu/substances-of-very-high-concern-identification-explained
Article 3(3) of the REACH Regulation defines an article as "an object which during production is given a special shape, surface or design which determines its function to a greater degree than its chemical composition". Chapter 2 of the Guidance on requirements for substances in articles provides information on how to determine if an object fulfils the above definition, including instructions on how to address borderline cases (Appendices 3 and 4).
The Guidance on requirements for substances in articles is available on the ECHA website at: http://echa.europa.eu/guidance-documents/guidance-on-reach
Packaging is considered a separate article under REACH. Please refer to section 2.5 of the Guidance on requirements for substance in articles for more information.
The Guidance on requirements for substances in articles is available on the ECHA website at: http://echa.europa.eu/guidance-documents/guidance-on-reach
There is no fee charged for the notification.
If the production/import ended before the substance was included in the Candidate List or before the notification obligation starts to apply (i.e. 6 months after a substance has been included in the Candidate List or 1 June 2011 for substances placed on the Candidate List before 1 December 2010) then you do not have to notify. Please note, however, that for the information requirements specified in Article 33 of the REACH Regulation, the date of supply of the article is the relevant date, i.e. these obligations also apply to producers and importers at the moment when they supply articles which were produced or imported before the substance was included in the Candidate List and are supplied after the inclusion. Please refer to section 3.2.1 of the Guidance on requirements for substances in articles for additional information.
The Guidance on requirements for substances in articles is available on the ECHA website at: http://echa.europa.eu/guidance-documents/guidance-on-reach
Once a substance enters the Candidate List you have to determine if you have the obligation to notify. One of the inputs needed to determine this is the tonnage of the substance in imported/produced articles per calendar year (or the average of the three previous years if the article has been imported/produced during three years before the start of the notification obligation). In this calculation, you have to include the tonnage in articles imported/produced for the full calendar year, i.e. if applicable, also before the date of the inclusion of the substance in the Candidate List. However, if the three year average is not available, the notifier will need to rely on the amounts of the previous calendar year.
The notification obligation also applies to producers or importers of articles containing recycled material. If you are a producer or importer, you must assess whether the articles you produce or import fall under the criteria of Article 7(2). It may be difficult to know the exact concentration of a Candidate List substance in, for example, recovered polymers where the concentration varies between each batch.
If your company concludes that the articles contain less than 0.1% of the Candidate List substance, we recommend you to document your basis for this conclusion in case of enforcement.
Yes. A non-EU producer of articles may appoint an Only Representative (OR) to submit a substance in articles notification.
The role and obligations of an Only Representative (OR) are explained in detail in chapter 2 of the Guidance on registration.
The Guidance on registration is available on the ECHA website at: http://echa.europa.eu/guidance-documents/guidance-on-reach
The notifications will not trigger a request to register the substance in articles, as such. However, the notification information can be used in addition to several other sources (e.g. registration information) to support identification of further needs for risk management.
If there are grounds for suspecting that the substance is released from the articles under normal or reasonably foreseeable conditions of use and such a release presents a risk to human health or the environment, you may be required as a producer or importer of articles to submit a registration. These decisions will be taken on a case-by-case basis and are not restricted to Candidate List substances.
Enforcement of the REACH Regulation, including the communication and notification obligations of Candidate List substances in articles, is in the remit of the authorities of the individual Member States.
You can find the contact details at: http://echa.europa.eu/support/helpdesks/national-helpdesks/list-of-national-helpdesks.
You should contact your national REACH helpdesk: http://echa.europa.eu/support/helpdesks/national-helpdesks
If you have questions on technical issues on REACH-IT, the link is available at: http://echa.europa.eu/support/dossier-submission-tools/reach-it and for questions related to IUCLID 6 at: http://iuclid.eu/ or if you are not in the European Economic Area (EEA), you may contact the ECHA Helpdesk: http://echa.europa.eu/contact
If you would like to check your general obligations under the REACH Regulation and how to fulfil them, you should use the Navigator: http://echa.europa.eu/support/guidance-on-reach-and-clp-implementation/identify-your-obligations.
For general guidance on the provisions of REACH that apply to substances in articles, you should consult the Guidance on requirements for substances in articles available at: http://echa.europa.eu/guidance-documents/guidance-on-reach.
For guidance on how to submit a notification (Article 7(2) of REACH), you should consult:
- the "Notifying substances in articles" page: http://echa.europa.eu/support/dossier-submission-tools/reach-it/notifying-substances-in-articles
- the Manual "How to prepare a substance in articles notification" if you are preparing an IUCLID dossier for your notification, but it also includes relevant advice and examples for all notifiers: http://echa.europa.eu/manuals
You are exempted from submitting a Candidate List substance in articles notification to ECHA if:
- you can exclude exposure of humans and the environment to the Candidate List substance in the articles during normal or reasonably foreseeable conditions of use, including disposal. Chapter 3.3.2 of the Guidance on requirements for substances in articles provides detailed information on the exemption based on “exclusion of exposure”
- the substance has already been registered for that use. Chapter 3.3.1 of the Guidance on requirements for substances in articles provides advice on how to find out if the substance is already registered for that use
The Guidance on registration is available on the ECHA website at: http://echa.europa.eu/guidance-documents/guidance-on-reach
If you conclude that you are exempted from notifying, in line with one of the two cases above, we recommend that you carefully document the basis for and the reasoning behind this conclusion. In this way, you will be prepared for any enforcement activities at national level.
Note that it might require more resources and be more difficult to properly assess and document exclusion of exposure or to find out if the substance is already registered for the use, than to prepare and submit a substance in articles notification.
Note that it may require more resources and be more difficult to demonstrate “no exposure” than to prepare and submit a substance in articles notification.
A producer/importer of an article wanting to demonstrate 'exclusion of exposure' has to ensure that the Candidate List substance does not come into contact with humans or the environment during the use, including disposal, of the article. Such a justification needs to show that no exposure to humans or the environment takes place during the article service life and the waste stage. For more information on “exclusion of exposure”-based exemption from notification, see chapter 3.3.2 of the Guidance on requirements for substances in articles at: http://echa.europa.eu/guidance-documents/guidance-on-reach
Note that it might require more resources and be more difficult to find out if the substance is already registered for the use, than to prepare and submit a substance in articles notification.
A substance can be considered as having already been registered for a particular use, if two conditions are fulfilled:
- the substance in question is the same as the substance already registered; and
- the use in question is the same as the use described in a registration of this substance, i.e. the registration refers to the use in the article.
In this context, “use” includes the use of the substance in the production of an article and, after being incorporated into the article, the use of the substance in the article during the article’s service life stages, including the waste stage.
For more information, see chapter 3.3.1 of the Guidance on requirements for substances in articles.
ECHA’s dissemination portal for substance information, which can be accessed via the ECHA website: https://echa.europa.eu/information-on-chemicals, contains information on registered substances provided by companies in their registration dossiers. It includes a variety of information on the substances which companies manufacture or import and may include information on the uses of the substance, unless the companies have claimed this information as confidential, including use of the substance in articles. The description of the use available here for all life cycle steps consists mainly of elements of the use descriptor system, as well as use name and in some cases contributing activity names. The information will normally not be sufficient on its own to conclude on the sameness of two uses for the purpose of establishing whether an exemption on the basis of Article 7(6) applies. Therefore, the use in question has to be described more in detail than just by using elements of the use descriptor system. For example, the published information that a substance has been registered for use in the Article Category 'Plastic articles' does not necessarily mean the registration is made to cover all plastic articles and all plastic materials. It could mean that use of the substance in production of some specific plastic articles is covered and described in the registration, while other plastic articles are not covered and assessed. The uses of two very different plastic articles may lead to very different exposures to humans and the environment. If the exposure related to the use of your article is not adequately assessed in a registration dossier, it cannot be considered a registered use.
Please note that there are limited possibilities to include information in section 3.5 of an IUCLID registration dossier, apart from the use descriptors. Section 3.5 of the IUCLID registration dossier may however contain 'free text' information, which is not based on the use descriptor system. Whether such information is sufficient to conclude on the sameness of use has to be examined on a case-by-case basis.
Most producers of articles are also downstream users under the REACH Regulation and as such have certain obligations outlined in Title V of the REACH Regulation. Since most substances on the Candidate List are already registered, producers of articles should already have communicated their use to the registrant for the purpose of registrations. Producers of articles may therefore not have to notify if their communicated uses are covered in the registration dossier.
Importers of articles may not have access to detailed information on registered uses. If you are not certain that your specific use is already registered, you should notify.
The Guidance on requirements for substances in articles is available on the ECHA website at: http://echa.europa.eu/guidance-documents/guidance-on-reach
Notification requirements apply only to substances already included in the Candidate List and meeting the criteria in Article 7(2) of the REACH Regulation. Therefore, if a substance is not yet in this list there is no need to notify. However, please note that CMR/PBT/vPvB and substances of equivalent concern that fulfil the criteria of Article 57 of the REACH Regulation can be included in the Candidate List. It is advised to keep track of the use of these substances in your articles and to follow the development of the Candidate List via the Registry of Intentions. By signing up for the ECHA e-News (http://echa.europa.eu/news-and-events/news-alerts) you will be alerted every time the Candidate List (http://echa.europa.eu/candidate-list-table) or Registry of Intentions (http://echa.europa.eu/addressing-chemicals-of-concern/registry-of-intentions) is updated with new substances.
One of the conditions that triggers your obligation to notify is when the concentration of the Candidate List substance exceeds 0.1% w/w in the article.
You should calculate the concentration of the substance for each article as produced or imported. This threshold applies to each article of an imported object made up of more than one article, which were joined or assembled together.
For more information and examples, see chapter 3.2.3.1 of the Guidance on requirements for substances in articles.
The Guidance on requirements for substances in articles is available on the ECHA website at: http://echa.europa.eu/guidance-documents/guidance-on-reach
One of the conditions that triggers the obligation for you to notify is when the total amount of the Candidate List substance present in all articles produced and/or imported exceeds one tonne per actor per year.
The articles concerned should contain more than 0.1% w/w of the substance. If an article contains the substance at a concentration below 0.1% w/w, this article does not have to be included in the tonnage calculation.
The calculation of the total amount in tonnes of the same Candidate List substance in all articles produced or imported (either isolated or incorporated in complex objects) by the same actor requires 3 steps:
1. Determination on whether the Candidate List substance in question is present at above the 0.1% w/w concentration threshold for each article produced or imported.
The calculation of the concentration of the Candidate List substances in articles or complex objects is done as described in Table 5 of the Guidance on requirements for substances in articles.
2. Calculate the amount in tonnes of the Candidate List substance in each article or article type produced or imported per year where it is present above the 0.1% w/w concentration threshold.
3. Calculate the total amount in tonnes for all articles by summing up the amounts calculated for each article or article type according to point 2 above.
The one tonne per year limit applies to the total tonnage produced/imported, in the European Economic Area (EEA) by the same legal entity.
If you believe you are close to one of the thresholds but are unsure whether you exceed it or not, we recommend you to notify.
Chapter 3.2.3.2 of the Guidance on requirements for substances in articles provides further explanations, namely on the concept of article type, and examples on how to calculate the tonnage.
The Guidance on requirements for substances in articles is available on the ECHA website at: http://echa.europa.eu/guidance-documents/guidance-on-reach
Identifying substances in articles, namely Candidate List substances, and quantifying their amounts is in many cases only possible if the respective information is made available by the actors in the supply chain. Supply chain communication is therefore the most important and efficient way of gathering the information needed in order to identify your obligations under REACH. This information can be obtained via, e.g.:
- Standardised REACH information from suppliers in the EU, e.g. safety data sheets (SDSs)
- Voluntary information tools to exchange information on articles, e.g. IT systems and tools
- Requesting information up the supply chain
It is advised that the information received from suppliers is properly evaluated.
The information you need can often be derived from standardised information that is obtained from suppliers of substances/mixtures based in the European Economic Area - EEA (e.g. SDSs or, where a SDS is not required, safety information and regulatory requirements (Article 32 of the REACH)). In the EEA, suppliers of articles containing more than 0.1% w/w of a Candidate List substance must provide available and relevant safety information (Article 33 of the REACH), including, as a minimum, the name of that substance.
Please be aware that the communication obligations arise from the presence of the Candidate List substance in the article. These obligations apply regardless of whether or not the supplier is aware of the presence of the substances. Therefore, it is in the interests of the supplier to seek information on the presence of Candidate List substances. Proactive requests in the supply chain are often useful to obtain the necessary information, in particular when the supplier of the article is outside the EEA.
Chemical analysis, although a possible way to identify and quantify substances in articles, is time consuming, costly and difficult to organise. If other approaches to obtaining information fail or become too complicated, conducting chemical analysis may nevertheless be an option to obtain information on the composition of articles. Chemical analysis may be helpful in certain situations. It can serve to obtain information needed for compliance with REACH and to confirm the information received from suppliers.
For more information, see Chapter 5 and Appendix 5 of the Guidance on requirements for substances in articles:
http://echa.europa.eu/guidance-documents/guidance-on-reach
The obligation to notify under Art. 7(2) of REACH and to communicate down the supply chain under Art. 33 of REACH only applies to articles which contain Candidate List substances.
Certain boron substances included in the Candidate List, such as diboron trioxide, boric acid and disodium tetraborate, are involved in processes leading to the production of articles containing “borosilicate glass”. In these processes, the boron substances are usually first chemically transformed into a manufactured glass substance. The glass substance is subsequently processed into articles. In these usual cases, the boron substances are completely transformed and are not present as such in the final glass article. Consequently, there is no obligation to notify under Art. 7(2) of REACH, nor to communicate information down the supply chain under Art. 33 of REACH.
Please note that it remains the responsibility of companies to assess for their specific use of the Candidate List boron substances whether these are completely transformed into glass in the manufacture of “borosilicate glass” and whether the Candidate List substance is present in the boron glass articles.
This information should be provided in your substance in articles notification. As a minimum, it should include the substance name and/or EC and CAS numbers.
You can find information on substance identity in the Candidate List available at: http://echa.europa.eu/candidate-list-table
Substance identity information, namely for group entries, may also be available in the "supporting documentation column" of the same webpage.
If you decide to prepare your notification online in REACH-IT, you only need to select your substance/Candidate List entry by typing the name, EC or CAS number in the correspondent field in the substance identification section of the wizard.
If you decide to prepare your notification beforehand in a IUCLID dossier and then upload it to REACH-IT, to facilitate the submission process, ECHA publishes pre-filled substance datasets with substance identity information in the Candidate List. The manual "How to prepare a substance in articles notification" provides guidance on how to download and use these datasets:
http://echa.europa.eu/manuals
This information should be provided in your substance in articles notification.
If you decide to prepare your notification online in REACH-IT, the classification is pre-filled automatically by the wizard for your selected Candidate List substance/entry. The classification is pre-filled for your Candidate List substance/entry as follows:
- If there is a match with an entry of Annex VI to CLP, the harmonised classification is included in your notification.
- If there is no match with an entry of Annex VI to CLP, the following information is included in your notification: “No harmonised classification according to Annex VI of CLP is available for this Candidate List entry”.
- If there are two or more matches with the entries of Annex VI to CLP, you need to select one; the selected harmonised classification entry is included in your notification.
If you decide to prepare your notification beforehand in a IUCLID dossier and then upload it to REACH-IT, to facilitate the submission process, ECHA publishes pre-filled substance datasets with classification information in the Candidate List. The manual "How to prepare a substance in articles notification" provides guidance on how to download and use these datasets:
/manuals
The Candidate List can be found at: /candidate-list-table
This information should be provided in your substance in articles notification by using the technical function descriptor, i.e. indicating the function of the Candidate List substance in the article, i.e. the role that the substance fulfils when it is used in the article (what it actually does in your article or in a mixture incorporated in the article).
If you decide to prepare your notification online in REACH-IT, in the Article(s)/Use(s) section of the wizard, you can describe the use(s) of the substance in the article(s) by using the “Technical function of the substance during use” field in each service life record.
If you decide to prepare your notification beforehand in a IUCLID dossier and then upload it to REACH-IT, you can describe the use(s) of the substance in the article(s) by using the "Technical function of the substance during use" field in section 3.5.6 of IUCLID. Chapter 7.3.3.1. of the manual "How to prepare a substance in articles notification" provides guidance on how to fill this IUCLID section.
Further details on the use(s) of the substance in the article(s) can be provided in the free text fields “Further description of use” of the on-line wizard or IUCLID, in the sections mentioned above, or if appropriate, by using other use descriptors (e.g. Process categories - PROC).
The manual "How to prepare a substance in articles notification" is available on the ECHA website at:
https://echa.europa.eu/manuals
Further information on the use descriptors can be found on the Guidance on Information Requirements and Chemical Safety Assessment - Chapter R.12 on use description available on the ECHA website at: https://echa.europa.eu/documents/10162/13632/information_requirements_r12_en.pdf/ea8fa5a6-6ba1-47f4-9e47-c7216e180197
In your notification you should identify all articles containing the Candidate List substance and provide for each one of them:
- a description of the article and its different integral parts (e.g. coating);
- a description where in the article the Candidate List substance is present, and if applicable, a description of the complex object where the article is joined or assembled as a component;
- if the article is to be used by consumers and/or workers; and
- a brief description of the use(s) of the article, including disposal.
The brief description of the use of the article during its service life, including disposal, should be provided by using relevant use descriptors as explained in detail in the Chapter R.12 on use description of the Guidance on Information Requirements and Chemical Safety Assessment. For articles the relevant descriptors are the following:
- Article category (AC) related to service life of articles containing the substance (and waste stage)
- Environmental release category (ERC)
- Process category (PROC) for production of articles and their use by workers
- Technical function (TF) of the substance in the article
You should also provide exposure-related information when describing the use(s) of your articles, including disposal. You may also wish to include information on the substance tonnage present in the article(s) and on the safe and proper use/handling of the article(s), as well as appropriate instructions for its disposal that you communicate under REACH Article 33.
Similar articles with similar uses can be grouped under the same use description.
The description of the use(s) of your article(s) must be provided in each created service life record created in the
- section Article(s)/Use(s) of the wizard, if you decide to prepare your notification online in REACH-IT, following the instructions in the help text; or
- section 3.5.6 of IUCLID, if you decide to prepare your notification beforehand in a IUCLID dossier and then upload it to REACH-IT; chapter 7.3.3.1. of the manual "How to prepare a substance in articles notification" provides guidance on how to fill this IUCLID section.
The manual "How to prepare a substance in articles notification", which also includes useful advice and examples for an online notification preparation, is available on the ECHA website at:
https://echa.europa.eu/manuals
Further information on the use descriptors can be found on the Guidance on Information Requirements and Chemical Safety Assessment - Chapter R.12 on use description available on the ECHA website at: https://echa.europa.eu/documents/10162/13632/information_requirements_r12_en.pdf/ea8fa5a6-6ba1-47f4-9e47-c7216e180197
This information should be provided in your substance in articles notification. As a minimum requirement, the tonnage range is to be provided.
If you decide to prepare your notification online in REACH-IT, in the section Tonnage, you must report the tonnage band of the substance contained in the articles and if you wish provide the actual estimated quantities. The help text of the wizard provides you guidance on how to fill this section.
If you decide to prepare your notification beforehand in a IUCLID dossier, you are required to report the tonnage band of the substance contained in the articles in the IUCLID Dossier Header, when creating the dossier to be uploaded to REACH-IT. In section 3.2 of IUCLID, you can indicate the tonnage of the Candidate List substance contained in the produced/imported articles. Chapters 7.3.1. and 8 of the manual "How to prepare a substance in articles notification" provides guidance on how to fill these IUCLID sections, including which years to base the calculation on.
You have the possibility to provide the tonnage of substance per article/use if you so wish in each created service life record created in the section Article(s)/Use(s) of the online wizard or in section 3.5.6 of IUCLID.
The manual "How to prepare a substance in articles notification" is available on the ECHA website at:
https://echa.europa.eu/manuals
See also Q&A 536
Only producers of articles who produce their articles in the European Economic Area (EEA) will have to enter their production sites. Article importers do not need to fill in this information.
If your company has several sites producing articles, you can list all the sites in one notification. Different notifications should not be made for different sites if the sites belong to the same legal entity.
If you decide to prepare your notification online in REACH-IT, please provide information on your production sites in section Role(s) in the supply chain after reporting your role as producer of article by selecting “Manufacturer”. Please refer to the help text of the online wizard for additional information.
If you decide to prepare your notification beforehand in a IUCLID dossier and then upload it to REACH-IT, please refer to chapters 3 and 7.3.2 of the manual "How to prepare a substance in articles notification" for additional information.
The manual "How to prepare a substance in articles notification" is available on the ECHA website at:
https://echa.europa.eu/manuals
This question is not relevant to notifications prepared online in REACH-IT.
To facilitate the submission of substance in articles notifications ECHA has made available pre-filled substance datasets in IUCLID format (i6z files) for substances on the Candidate List. These datasets are only relevant if you decide to prepare your notification beforehand in a IUCLID dossier and then upload it to REACH-IT. They contain information on substance identification, composition and classification and labelling. Each substance has its own pre-filled dataset, which can be downloaded from the Candidate List webpage.
Chapters 5 and 6 of the manual "How to prepare a substance in articles notification" provide information on how to download and use the datasets. This manual is available on the ECHA website at:
https://echa.europa.eu/manuals
The Candidate List is published on ECHA's website at: http://echa.europa.eu/candidate-list-table
The registration number only needs to be included in the notification dossier if the substance has already been registered and the registration number is available to the notifier. If the article is imported from outside the EU, this field does not need to be filled. Producers of articles, incorporating the Candidate List substance into the article themselves, should normally have access to the registration number via the Safety Data Sheet (SDS) for the substance. In this case, the article producer is likely to be exempt from the notification obligation however he should verify that his use is indeed covered by the registration (see Q&A 530).
If available, the registration number should be provided in your substance in articles notification.
It can be inserted in the
- section Administrative information of the wizard, if you decide to prepare your notification online in REACH-IT, following the instructions in the help text; or
- section 1.3 of IUCLID, if you decide to prepare your notification beforehand in a IUCLID dossier and then upload it to REACH-IT; chapter 7.1.3. of the manual "How to prepare a substance in articles notification" provides guidance on how to enter this information in IUCLID. This manual is available on the ECHA website at:
https://echa.europa.eu/manuals
Article 3(30) of the REACH Regulation specifies that "per year" means "per calendar year", i.e. 1st January – 31st December. However, for the purpose of the tonnage calculation, if the article has been manufactured or imported for at least three consecutive years the average volume of the preceding three years is recommended to be used. However, if the three year average is not available, the notifier will need to rely on the amounts of the previous calendar year.
Notification is required from producers or importers of articles, therefore you have to notify only when the object manufactured/imported is an article. Please refer to Chapter 2 of the Guidance on requirements for substances in articles for additional information on the distinction between a substance/mixture and an article. You should also refer to Tables 3 and 4 of the Guidance, in chapter 3.2.2, if you are dealing with coated articles and complex objects.
The Guidance on requirements for substances in articles is available on the ECHA website at: http://echa.europa.eu/guidance-documents/guidance-on-reach
You have to submit your substance in articles notification via REACH-IT. Therefore you must have an active REACH-IT account to be able to submit your notification.
Before submission, there are two possibilities to prepare your notification:
- Preparing your notification online in REACH-IT
This easy-to-use method is recommended for all notification submitters, especially those who are not familiar with IUCLID. All instructions and information requirements to carry out the notification are provided at every step. - Preparing and uploading a IUCLID dossier to REACH-IT
This method is recommended for notification submitters who are already users of IUCLID, as well as for those who also want to report different information, from that pre-filled in the online wizard in REACH-IT, on substance identification, composition, or classification and labelling.
Please use this method, if you wish to claim some of the notified information as confidential, in addition to information which is always treated as confidential (see Article 118 of REACH).
To prepare your IUCLID dossier, you can find a detailed description in the manual “How to prepare a substance in articles notification”, which is available on the ECHA website at: https://echa.europa.eu/manuals
You must have an active REACH-IT account to be able to submit your notification.
- Log in to REACH-IT and select “Submit a dossier” (landing page or menu)
- Select from the list of dossier types “Substance in articles”
- Select either to upload a IUCLID dossier, or to prepare your notification online in REACH-IT
- Submit your notification by following the wizard instructions
Once your notification has been submitted in REACH-IT, you can follow its submission status using the submissions or substances pages in REACH-IT.
The update of your notification needs to be initiated from the reference number page in REACH-IT.
Please find more information on ECHA website at https://www.echa.europa.eu/support/dossier-submission-tools/reach-it/notifying-substances-in-articles
A notification of a substance in articles shall be made at the latest 6 months after the substance has been included on the Candidate List. The obligation started to apply from 1 June 2011.
If you have the obligation to notify a substance in your articles and the deadline has already passed, you are encouraged to notify immediately. You might face national enforcement sanctions if you fail to meet the deadline. Although ECHA accepts notifications after the legal deadline, this does not prevent your national enforcement authorities to impose sanctions on you.
If the import of the substance in articles starts after the notification deadline has expired (i.e. 6 months after the inclusion in the Candidate List), notifications must be made without undue delay as soon as the conditions related to the notification obligation are met, i.e. as soon as the 1 tonne per year threshold has been reached.
One notification should be submitted for all the articles produced or imported containing the same substance by/to one legal entity. You are however requested to indicate the uses for each different type of article in your notification.
If you are producing or importing new articles containing the Candidate List substance above 0.1% w/w, which are not reported in your submitted notification, you must update your notification without undue delay, given that the conditions related to the notification obligation are met.
No, it is necessary to submit a separate notification for each substance and for each importer/manufacturer.
In order to be able to demonstrate compliance with your obligations, you are recommended to update your notification if the information included in the notification changes. Examples of important changes could be: change in tonnage range.
However, if you are producing or importing new articles containing the Candidate List substance above 0.1% w/w, which are not reported in your submitted notification, you must update your notification without undue delay, given that the conditions related to the notification obligation are met.
The update of your notification needs to be initiated from the reference number page in REACH-IT. You can access to this page by log in to your REACH-IT account at https://idp-industry.echa.europa.eu/idp/
At the top of the reference number page in REACH-IT, you can update your notification by clicking on
- “Create and submit online an update”, if you have prepared your notification online in REACH-IT or you have submitted it via web form (closed since October 2017); To prepare and submit your notification update online, please follow the wizard instructions.
- “Submit a IUCLID dossier update”, if you have submitted your notification by uploading a IUCLID dossier; After updating your IUCLID dataset and creating your updated notification IUCLID dossier, you can submit the dossier update via REACH-IT, by uploading it; Please follow the submission wizard instructions.
If you stop importing or producing articles containing a Candidate list substance, you should inform ECHA directly via REACH-IT using the "Cease manufacture" functionality in the reference number page.
Please find further guidance on REACH-IT at https://www.echa.europa.eu/support/dossier-submission-tools/reach-it
Under some circumstances, article importers have to register or notify substances in articles to ECHA (see Article 7 of REACH): these obligations are in general the same as for producers of articles. When placing articles on the market in the EEA, importers of articles may also have to communicate information on substances in their articles to their customers. In order to establish whether registration, notification or communication duties apply, any importer of articles is advised to follow first the Guidance in a Nutshell on requirements for substances in articles.
Substances may be intended to be released from articles to provide "added value". Scented children's toys, for example, are articles made with the intent of releasing substances. The release is an additional quality of the toy and is therefore intended because it gives added value, namely a pleasant smell.
As a counter-example, consider the case of a printer cartridge or a wet cleaning wipe. In these cases, the substances can be physically separated from the article. Therefore, they cannot be considered as substances in an article, but rather as substances in a container.
The consequence is that the supplier of cartridges is considered as a supplier of substances and the general registration obligations therefore apply.
For further information see the Guidance on requirements for substances in articles:
In most cases, if you want to find out for which uses a substance has been registered, you will have to ask other actors up your supply chain.
For this purpose, you need to describe the function of the substance in the article, the process by which the substance is included in the article and into which type of article. This description should be in line with the use descriptor system.
Safety data sheets (SDSs) can be helpful as they contain information on uses of the substance or mixture as far as they are known by the supplier. If the SDS also includes a registration number, it may be possible, depending on the accuracy of the use descriptions in the SDS, to conclude that a particular use of this substance has already been registered. However, if you have doubts, you should seek confirmation from the actual registrant up the supply chain.
Alternatively, you could identify and ask manufacturers or importers of that substance from any supply chain for the uses they have registered this substance for, or whether they have registered it for a particular use.
A good way to identify manufacturers and importers of a substance is to launch a corresponding request within the substance information exchange forum (SIEF) for this substance, provided that you have pre-registered the substance or joined the SIEF as a data holder.
For further information see the Guidance on requirements for substances in articles:
Yes, it is.
Entry 3, first paragraph, states that for an ornamental lamp to be within the scope of the restriction, light or colour effects need to be produced by means of different phases. The restriction entry does not define the term ‘different phases’.
However, the original intention of Council Directive 79/663/EEC of 24 July 1979 that introduced this entry was to prevent harm caused by the breakage of ornamental objects and the release of hazardous substances. Since the liquid used in the LED lamp is classified as Skin irritant, H319, it falls within the scope of paragraph (b) in column 1 of entry 3, and thus the product in question is within the scope of the restriction.